Sumario
Principales motivos de consulta
2017, vol. 10, nº 4


 Retraso del desarrollo psicomotor. Fundamentos diagnósticos
Retraso del desarrollo psicomotor. Fundamentos diagnósticos
Autores: García Pérez A1, García Cruz JM2
1 Sociedad Española de Neurología Infantil. Neuropediatra. Hospital Universitario Fundación Alcorcón. Madrid (España).
2 Sociedad Española de Neurología Infantil. Neuropediatra. Hospital Universitario Fundación Alcorcón. Madrid (España).
1 Sociedad Española de Neurología Infantil. Neuropediatra. Hospital Universitario Fundación Alcorcón. Madrid (España).
2 Sociedad Española de Neurología Infantil. Neuropediatra. Hospital Universitario Fundación Alcorcón. Madrid (España).
PUNTOS CLAVE
- El retraso psicomotor (RPM) es un rendimiento menor a dos desviaciones estándar en al menos dos escalas: motora (gruesa/fina), lenguaje, social y habilidades de la vida diaria.
- Usamos este término en los menores de 5 años y por encima de esta edad el de retraso mental (RM) o discapacidad intelectual (DI).
- El RPM está incluido en la quinta edición del Manual diagnóstico y estadístico de los trastornos mentales (DSM-5) y en la décima edición de la clasificación internacional de enfermedades (CIE-10), en los trastornos del desarrollo o del neurodesarrollo.
- La detección de signos de alarma por parte del pediatra de Atención Primaria es muy importante en el programa de seguimiento del niño sano o programa de salud infantil (PSI).
- Para ello se utilizan test de cribado o screening (Denver y Haizea-Llevant) y escalas del desarrollo.
- Los estudios genéticos, metabólicos y neurorradiológicos son los test de primera línea en la investigación etiológica del RPM.
- Llegar a un diagnóstico es importante para proporcionar un tratamiento, si lo hay, para dar un pronóstico y administrar un consejo genético que evite la recurrencia en las familias afectadas.
- El tratamiento preferible es etiológico, pero no siempre es posible.
- Habitualmente se realiza un abordaje funcional con medidas de atención temprana y educativas.
INTRODUCCIÓN
El retraso psicomotor (RPM) o global developmental delay es para la Child Neurology Society un rendimiento menor a dos desviaciones estándar (DE) en al menos dos escalas: motora (gruesa/fina), lenguaje, social y habilidades de la vida diaria; es un trastorno de inicio en la infancia y con curso evolutivo estable1. Usamos este término en los menores de 5 años y por encima de esta edad el de retraso mental (RM) o discapacidad intelectual (DI). El RPM está incluido en la DSM-5 y la CIE-10 en los trastornos del desarrollo o del neurodesarrollo, que corresponden al grupo de condiciones con inicio en el periodo de desarrollo y se manifiestan generalmente antes de entrar en la escuela.
La prevalencia del RPM es del 2,5-3%, pero no siempre predice un RM, pues algunos mejoran o desarrollan sus capacidades potenciales. La prevalecía es mayor en varones (1,5:1) y en niveles socioeconómicos bajos/educación limitada. El RPM genera importantes costes familiares, a los sistemas de salud y educativos2,3.
Son signos de alarma en el desarrollo psicomotor un retraso significativo en la aparición de adquisiciones globales del desarrollo o de adquisiciones en algún área específica. También lo son la persistencia de patrones que deberían haber desaparecido (por ejemplo, reflejos arcaicos) o de conductas anormales a partir de una edad (por ejemplo, movimientos repetitivos en mayores de 8 meses), y por supuesto la existencia de signos anómalos a cualquier edad (por ejemplo, movimientos oculares anormales). Un signo de alarma no presupone un problema, pero obliga a un examen y seguimiento.
La detección de estos signos de alarma por parte del pediatra es muy importante en el programa de seguimiento del niño sano o programa de salud infantil (PSI). No obstante, la impresión subjetiva en la evaluación del RPM es insuficiente. Es importante utilizar test de cribado o screening (Denver y Haizea-Llevant) y escalas del desarrollo (Bayley, Batelle, McCarthy…), que sistematizan la exploración, evitan dejar de valorar algún aspecto y aumentan la detección de los trastornos. Las escalas del desarrollo dan un cociente de desarrollo (CD) que alerta cuando no es satisfactorio, aunque su poder predictivo del cociente intelectual (CI) futuro es escaso4 (Tabla 1).
Tabla 1. Escalas de evaluación del desarrollo. Mostrar/ocultar
El retraso no sindrómico se define como aquel sin ninguna otra característica clínica que lo haga discernible. Determinar la etiología en estos casos es un reto para el pediatra (Tabla 2). Un alto número de RPM se pueden atribuir a causa genética (30-40%) y errores congénitos del metabolismo (ECM) (1-5%). Actualmente unos 450 genes están implicados en el RPM; de ellos, 400 se relacionan con retraso sindrómico y 50 con retraso no sindrómico. Además, aproximadamente un 50% de los 600 ECM conocidos conllevan retraso del desarrollo y de ellos un importante número son susceptibles de tratamiento. Los estudios genéticos, metabólicos y neurorradiológicos son los test de primera línea en la investigación etiológica del RPM5,6.
Tabla 2. Etiología del retraso psicomotor/discapacidad intelectual. Mostrar/ocultar
Llegar a un diagnóstico es importante para dar un pronóstico, administrar un consejo genético que evite la recurrencia en las familias afectadas, poder dar los tratamientos disponibles, evitar más pruebas (biopsias de piel o músculo, pruebas neurofisiológicas…), manejar la comorbilidad acompañante y dar soporte a las familias con la información específica7.
En base a la anamnesis y a la exploración se establece la etiología del RPM en un 17,2-34,5% de casos, y obtenemos claves para el diagnóstico en un 62-79%. Cuando exista una hipótesis diagnóstica, se realizarán técnicas dirigidas a confirmar la presunción clínica (genéticas, pruebas metabólicas, de neuroimagen o neurofisiológicas). En los casos no sindrómicos se deben realizar pruebas de menor a mayor complejidad y coste para identificar una etiología. Si un tercio de los casos se diagnostican por la anamnesis y el examen físico, otro tercio es por estudios complementarios basados en la hipótesis diagnóstica, y en el tercio restante por pruebas de barrido sin sospecha diagnóstica2,6.
HISTORIA CLÍNICA Y EXPLORACIÓN FÍSICA
Anamnesis e historia clínica
Deben recoger:
- La edad de inicio del RPM (de 1-12 meses, de 1-5 años, de 5-15 años)8.
- Prematuridad, crecimiento intrauterino restringido (CIR), factores de riesgo neurológico o psicosocial (Tabla 3 y 4), abortos, fetos muertos, muertes neonatales o infantiles… en familiares de primer y segundo grado, consanguineidad o etnias más endogámicas, otros familiares afectos…
- En varones, preguntar a lo largo de tres generaciones por hombres afectados en la rama materna, lo que indicaría herencia ligada al X.
- Factores gestacionales prenatales: edad materna y paterna al nacimiento, técnicas de fertilidad, alcohol (niños adoptados), drogas, hidantoínas, valproato…, exposición a radiación (especialmente entre 9-15 semanas de gestación), malnutrición materna, hiperfenilalaninemia materna o hipotiroidismo durante la gestación, diabetes mellitus materna… La autodeclaración del consumo de tóxicos en la gestación es inferior a un tercio de las ocasiones que tiene lugar9,10.
- Curso clínico: evolución hasta el momento de todas las áreas de desarrollo psicomotor, descompensaciones episódicas, regresión…
- Cribado endocrinometabólico (lugar y fecha de nacimiento). Actualmente, existen diferencias entre los programas de cribado neonatal de las distintas comunidades autónomas (CC. AA.). A pesar de estar establecidas el mínimo de enfermedades a cribar: hipotiroidismo congénito, fenilcetonuria, fibrosis quística, déficit de acil-coenzima A deshidrogenasa de cadena media (MCADD), deficiencia de 3-hidroxi acil-coenzima A deshidrogenasa de cadena larga (LCHADD), acidemia glutárica tipo 1 y anemia falciforme; solo el hipotiroidismo congénito y la fenilcetonuria son detectadas por todas las CC. AA. Sigue siendo necesaria la creación de un programa de cribado neonatal único a nivel estatal.
Tabla 3. Factores de riesgo neurológico. Mostrar/ocultar
Tabla 4. Factores de riesgo psicosocial. Mostrar/ocultar
Algunas de las siguientes enfermedades son cribadas según la CC. AA.11:
- Hiperplasia adrenal congénita.
- Anemia falciforme.
- Aminoacidopatías: tirosinemias, enfermedad de orina con olor a jarabe de arce (MSUD), homocistinuria, argininemia, citrulinemia 1 y 2 y aciduria argininosuccínica.
- Enfermedades del metabolismo de la β-oxidación de ácidos grasos (AG): deficiencia de acil-coenzima A deshidrogenasa de cadena corta (SCADD), deficiencia de acil-coenzima A deshidrogrenasa de cadena muy larga (VLCADD), deficiencia múltiple de acil-coenzima A deshidrogenasas (MADD), deficiencia de carnitina palmitoiltransferasa I y II (CPT-I y CPT-II) y deficiencia en la captación celular de carnitina (CUD).
- Otras acidurias orgánicas (propiónica, metilmalónica, isovalérica), deficiencia múltiple de carboxilasas (deficiencia de holocarboxilasa sintetasa y de biotinidasa), metilcrotonilglicinuria y deficiencia de β-cetotiolasa.
Las CC. AA. que hacen un cribado neonatal más completo son Andalucía, Extremadura, Murcia, Melilla y Cataluña. Hacen cribado extendido aunque no tan amplio Aragón, La Rioja, Castilla-La Mancha, Madrid y Galicia. Las CC. AA. que menos determinan en su cribado son Asturias, Cantabria, Navarra, País Vasco, Castilla-León, Valencia, Baleares y Canarias.
Exploración
Debe recoger:
- Mímica y mirada, comportamiento, impresión del nivel intelectual y lenguaje (los retrasos leves pasan más desapercibidos y hay que hacer valoración de CD/CI).
- Somatometría (principalmente descartar micro- y macrocefalia).
- Dismorfias (pelo, cráneo, cara, dermatoglifos, extremidades).
- Disrafismos espinales.
- Discromías.
- Visceromegalias, cardiopatías, alteraciones genitourinarias. Cualquier signo que oriente a una etiología genética (Tabla 5).
- Examen visual y auditivo.
-
Examen neurológico:
-
Exploración motora:
-
Tono muscular:
- Tono pasivo (distensibilidad, pasividad, trofismo).
- Tono activo (respuesta a maniobras: tracción, Landau…).
- Rigidez, espasticidad.
- Pares craneales.
- Reflejos (reflejos osteotendinosos [ROT], superficiales –reflejo cutáneo-plantar [RCP]–, arcaicos).
- Fuerza.
- Movimientos anormales.
-
Tono muscular:
-
Función propioceptiva y cerebelosa:
- Equilibrio (Romberg, marcha).
- Coordinación y función motora fina (dedo-nariz, talón-rodilla).
-
Sensibilidad:
- Superficial (tacto, dolor, temperatura).
- Profunda (posicional y vibratoria).
- Discriminatoria (discriminación, estereognosia y grafestesia).
-
Exploración motora:
Tabla 5. Indicadores clínicos de etiología genética. Mostrar/ocultar
Si tras una anamnesis y exploración detenida no se llega a ningún diagnóstico de sospecha, pasaremos a las pruebas complementarias (Figura 1).
Figura 1. Algoritmo etiológico para el retraso mental y la discapacidad intelectual en niños. Atención especializada. Mostrar/ocultar
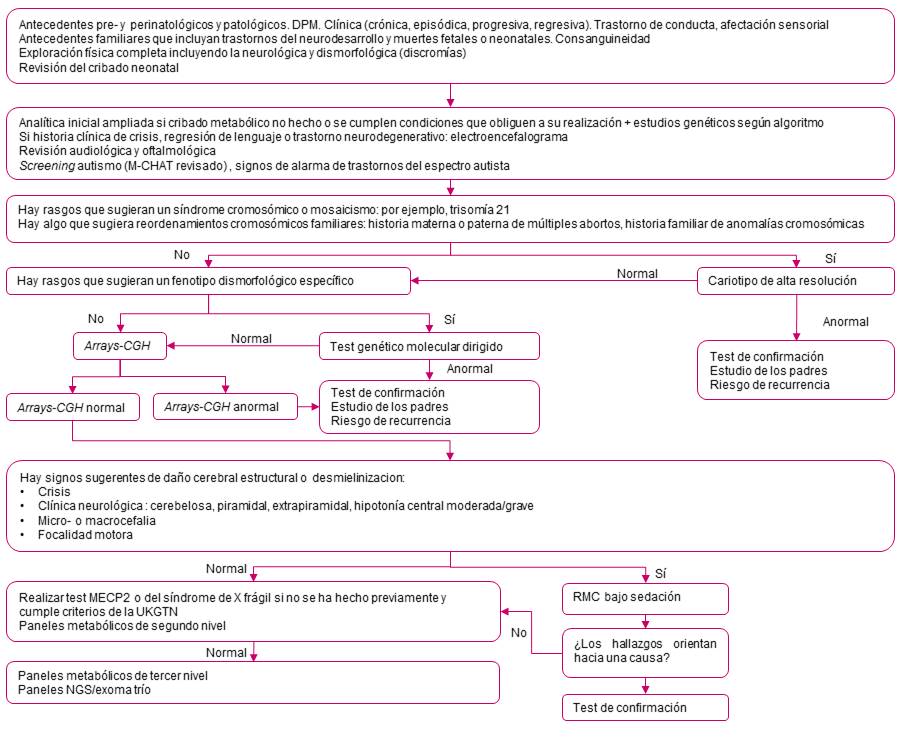
PRUEBAS COMPLEMENTARIAS
El rendimiento diagnóstico de las pruebas es de alrededor de un 20%5,11.
Pruebas genéticas
Tras obtener el consentimiento informado.
Array-Hibridación genómica comparada (array-CGH)
Son el gold standard del análisis genético de primera línea, reemplazando al cariotipo. Su rendimiento diagnóstico es mayor por su sensibilidad para deleciones y duplicaciones submicroscópicas, la identificación de genes específicos incluidos en las pérdidas o ganancia de material genético y la definición de los puntos de corte y tamaños exactos de los desequilibrios; en todo el genoma y con alto nivel de resolución. Los arrays no detectan mutaciones puntuales, ni mosaicismos genéticos inferiores al 20%, ni reordenamientos equilibrados como translocaciones recíprocas balanceadas. Se le considera el “cariotipo molecular”. El American College of Medical Genetics and Genomics actualizó sus guías hace unos años para recomendar los arrays como test de primera línea en la evaluación posnatal de pacientes con retraso, trastorno del espectro autista (TEA) y anomalías congénitas múltiples12.
Se pueden obtener tres resultados de los arrays: normal, anormal con una variante patogénica, anormal con variante de significado desconocida o incierta (variants of unknown significance [VUS]). Las VUS son con frecuencia variaciones en el número de copia (VNC) de ADN de ciertos segmentos del genoma, y que involucran de un nucleótido a 1000 pb (llamados polimorfismos). Cada persona tiene aproximadamente 100 VNC que afectan a numerosos genes y solo a veces son patogénicas. Es difícil establecer el significado de algunas VNC y se recomienda entonces realizar arrays-CGH en los padres como ayuda a la interpretación. Cuanto mayor sea el polimorfismo y más genes involucre es más probable que sea causal, aunque esto no siempre es verdad (variantes de más de 500 kb están presentes en el 5-10% de la población y superiores a 1 Mb en el 1-2%). Por otro lado, la diferente penetrancia de los mismos polimorfismos en distintos miembros de una familia requiere ser cautos en la interpretación.
Los arrays-CGH tienen un rendimiento del 15-20%, el test del síndrome de X frágil (SXF) de 0,6-1% y el cariotipo de alta resolución (bandas G) es de 1-3% (si se excluyen los Down y otros síndromes reconocibles). El coste-beneficio de los arrays, por su alto rendimiento diagnóstico, beneficia los costes del conjunto de pruebas etiológicas para el RPM. Es más rentable realizar un array-CGH de entrada que comenzar con cariotipo y estudios adicionales. Triplicar el número de diagnósticos (pasar del 7 al 22%) significa solo un incremento del 1% del coste por diagnóstico. Por ello, la sustitución de la citogenética por los arrays-CGH además de estar justificada por motivos científico/asistenciales, es irrelevante desde el punto de vista económico13-15.
Cariotipo de alta resolución (800 bandas)
No detecta anomalías menores a 5-10 Mb. Se recomienda como prueba de primera línea si:
- Se reconoce un síndrome tipo aneuploidía: trisomía 13, 18 o 21, monosomía X (Turner), trisomía X, XXY (Klinefelter), XYY (superhombre). Estos cuatro últimos no suelen conllevar retraso del desarrollo, pero sí, a veces, de lenguaje, de aprendizaje y conducta.
- Hay historia familiar de reordenamientos cromosómicos.
- Hay historia materna o paterna de abortos múltiples.
- Si se sospecha bajo grado de mosaicismo (< 20%) o de cromosomas en anillo.
El cariotipo en fibroblastos cultivados de biopsia de piel hipopigmentada en pacientes con discapacidad intelectual y discromías nos puede dar el diagnóstico de algún tipo de mosaicismo (por ejemplo, diploide/triploide)16.
Estudio del síndrome x-frágil (SFX)
Se recomienda realizar inicialmente este estudio molecular de la expansión del triplete CGG en el gen FMR1 (Xq27.3) en:
- Varones con discapacidad moderada/severa.
- Mujeres con discapacidad moderada/leve y si hay antecedentes de insuficiencia ovárica prematura (amenorrea en menores de 40 años)/FXPOI en portadoras de la premutación (expansión triplete CGG de 55-230).
- Varones con temblor-ataxia/FXTAS portadores de premutación.
La frecuencia del SFX es significativamente más baja que previamente. Su fenotipo variable hace que el diagnóstico de X frágil sea difícil. Las guías de la United Kingdom Genetic Testing Network for FXS Testing (UKGTN) aumentan 10 veces la eficacia de la genética molecular, sin perder casos17-19 (Tabla 6).
Tabla 6. Guías de la United Kingdom Genetic Testing Network para el síndrome de X frágil. Mostrar/ocultar
Otros test genéticos moleculares
Dirigidos a mutaciones, genes o regiones genómicas específicas. Ejemplos:
- Gen MECP2 para el síndrome de Rett (1,5% de niñas y 0,5% de varones con retraso moderado-severo).
- Hibridación fluorescente in situ (FISH) para microdeleciones en la región cromosómica 7q11.23 para el síndrome de Williams.
- Alteraciones de metilación de la región 15q11.2 para los síndromes de Angelman y Prader-Willi.
- Estudio molecular por reacción en cadena de la polimerasa (PCR) de la amplificación de repeticiones del triplete CTG en el locus 19q13-2 para la distrofia miotónica de Steinert.
Las pruebas FISH, PCR y multiplex ligation-dependent probe amplification (MLPA) identifican desequilibrios menores a 1 Mb. El MLPA es una PCR múltiple que permite más experimentos en una misma reacción, diseñándose kits para varias regiones genómicas vinculadas al retraso.
Secuenciación NGS (Next Generation Sequencing)
Se utiliza para el estudio de mutaciones puntuales asociadas al retraso. Para ello se realizan paneles NGS y exomas clínicos. Los primeros ofrecen mayor precisión diagnóstica frente a más datos genómicos del exoma clínico completo.
Se hacen paneles NGS en los siguientes casos:
- Si se requiere una alta precisión diagnóstica (fenotipo clínico característico).
- Sospecha de enfermedad asociada a genes de gran tamaño.
- Para enfermedades con heterogeneidad genética (diversos genes específicos).
- Enfermedades con signos clínicos comunes (miopatías congénitas).
Se hace exoma clínico con todos los genes del catálogo Online Mendelian Inheritance in Man (OMIN) para:
- Enfermedades con fenotipos no característicos y gran heterogeneidad genética (por ejemplo, TEA).
- Pacientes con más de un fenotipo característico (RPM/DI y TEA).
- Estudio de mutaciones de novo (hay que estudiar a los padres también).
- Casos de retraso con todas las pruebas genéticas negativas.
El análisis familiar utilizando tríos (hijo afectado y padres) parece emerger como la metodología de elección para detectar mutaciones puntuales de novo vinculadas al retraso del desarrollo o discapacidad intelectual20. El abaratamiento de costes de los exomas y paneles y la mejora del análisis de los datos, hacen que la secuenciación masiva llegue a ser la herramienta genética compañera de los arrays en el diagnóstico de niños con retraso.
Pruebas metabólicas
Los ECM más frecuentes que pueden determinar un retraso inespecífico son algunas aminoacidopatías como la fenilcetonuria y la MSUD, los trastornos del ciclo de la urea, la homocistinuria clásica (deficiencia de cistationina β-sintasa) y otros trastornos relacionados con el metabolismo de la homocisteína, el déficit de síntesis y transporte de creatina, las acidurias orgánicas como la aciduria glutárica, la enfermedad de San Filippo (MPS-III), el déficit de adenilosuccinato liasa, el síndrome de Lesch-Nyhan, los defectos congénitos de glicosilación, y más raramente la deficiencia de trasporte cerebral de glucosa (GLUT-1), los trastornos peroxisomales y los del metabolismo del cobre6.
Las pruebas metabólicas para el diagnóstico de un ECM como causa de RPM/DI tienen un rendimiento bajo (1-3%) en los países donde se realiza cribado neonatal, sin embargo, una revisión actual de la literatura médica6 ha identificado 89 ECM que se presentaron con RPM como característica prominente y además susceptibles de terapia.
El cribado metabólico inicial (analítica de primer nivel y segundo nivel en nuestro texto) es relativamente asequible, poco invasivo e identifica el 60% de los ECM susceptibles de tratamiento. El 40% restante se diagnostica con pruebas de barrido de tercer nivel21. Los síntomas y hallazgos del examen físico o de la neuroimagen sugestivos nos permitirán adelantar test metabólicos específicos según la sospecha clínica. Con esta orientación y protocolo el rendimiento de los exámenes metabólicos para el RPM aumenta al 5%.
El tratamiento temprano supone en los casos tratables una mejoría o estabilización del desarrollo psicomotor, conducta, crisis y de las manifestaciones neurológicas y sistémicas. La clave para pensar en un ECM como causa de un RPM/DI es la presencia de síntomas neurológicos, psiquiátricos y extraneurólogicos múltiples, los rasgos dismórficos y el curso progresivo-regresivo del proceso8.
Los estudios metabólicos no están indicados en el examen inicial del RPM, dados los programas de cribado. No obstante, el cribado metabólico neonatal solo detecta aproximadamente un 10% de todas las ECM, y además hay variantes de las enfermedades que se criban en dichos programas que no se detectan a los 2 días de vida, que es cuando se toma la muestra, y debutarán más tarde en el niño. El cribado neonatal es importantísimo, pero no es 100% efectivo ni siquiera para ese 10% de enfermedades que se criban. El que las pruebas metabólicas neonatales hayan sido normales no descarta al 100% un ECM y es obligado realizar pruebas metabólicas, ya inicialmente, si hay:
- Historia familiar positiva: abortos, fetos muertos, muertes neonatales o infantiles, niños con iguales problemas en familiares de primer o segundo grado, consanguineidad o pertenencia a etnias más endogámicas.
- Fallo de medro, olor especial, dieta selectiva…
- Deterioro o afectación progresiva cognitiva o motora.
- Rasgos toscos o dismórficos, macro- o microcefalia, organomegalias, afectación visual o auditiva, manifestaciones cutáneas, disfunción orgánica múltiple…
- Afectación neurológica: episodios encefalopáticos, crisis, ataxia, hipotonía-hipertonía, trastornos del movimiento (signos piramidales o extrapiramidales).
- Resultados analíticos o de neuroimagen que orienten hacia a algún ECM.
CUADERNO DEL PEDIATRA
- El RPM es un rendimiento menor a 2 DE en al menos dos escalas: motora (gruesa/fina), lenguaje, social y habilidades de la vida diaria; de inicio en la infancia y con curso evolutivo estable.
- Usamos este término en los menores de 5 años, y por encima de esta edad el de retraso mental (RM) o discapacidad intelectual (DI).
- El RPM está incluido en la DSM-5 y CIE-10 en los trastornos del desarrollo o del neurodesarrollo, que corresponden al grupo de trastornos con inicio en el periodo de desarrollo y se manifiestan generalmente antes de entrar en la escuela.
- La prevalencia del RPM es del 2,5-3%, mayor en varones y niveles socioeconómicos bajos/educación limitada.
BIBLIOGRAFÍA
- Fernández-Jaén A, Cigudosa JC, Martín Fernández-Mayoralas D, Suela J, Fernández-Perrone AL, Calleja-Pérez B, et al. Genética aplicada a la práctica clínica en los trastornos del neurodesarrollo. Rev Neurol. 2014;58:S65-S70.
- González G, Raggio V, Boidi M, Tapié A, Roche L. Avances en la identificación etiológica del retraso mental. Rev Neurol. 2013;57:75-83.
- Pivalizza P, Lanani SL. Intellectual disability in children: evaluation for a cause. En: UpToDate [en línea] [consultado el 02/11/2017]. Disponible: https://www.uptodate.com/contents/intellectual-disability-in-children-evaluation-for-a-cause
- García Pérez A, Martínez Granero MA, Sánchez Calderón M, Izquierdo López L. Retraso psicomotor y retraso mental. Clasificación y etiología. Valoración y manejo. En: Verdú A, García A, García O, Arriola G, Martínez B, de Castro P (eds.). Manual de Neuropediatría. 2.ª edición. Madrid: Panamericana, 2014. p. 331-44.
- O’Byrne JJ, Lynch SA, Treacy EP, King MD, Betts DR, Mayne PD, et al. Unexplained developmental delay/learning disability: guidelines for best practice protocol for first line assessment and genetic/metabolic/radiological investigations. Ir J Med Sci. 2016;185:241-8.
- Van Karnebeek CD, Shevell M, Zschocke J, Moeschler JB, Stockler S. The metabolic evaluation of the child with an intellectual developmental disorder: diagnostic algorithm for identification of treatable causes and new digital resource. Mol Genet Metab. 2014;111:428-38.
- McDonald L, Rennie A, Tolmie J, Galloway P, McWilliam R. Investigation of global developmental delay. Arch Dis Child. 2006;91:701-5.
- Saudubray JM, van den Berghe G, Walter JH (eds.). Inborn metabolic diseases: diagnosis and treatment. 5.ª edición. Berlín: Springer; 2012.
- Chaabane S, Sheehy O, Monnier P, Fraser W, Bissonnette F, Trasler JM, et al. Ovarian stimulation, intrauterine insemination, multiple pregnancy and major congenital malformations: a systematic review and meta- analysis- The ART_Rev Study. Curr Drug Saf. 2016;11:222-61.
- Julvez J, Álvarez-Pedrerol M, Rebagliato M, Murcia M, Forns J, García-Esteban R, et al. Thyroxine levels during pregnancy in healthy women and early child neurodevelopment. Epidemiology. 2013;24:150-7.
- Srour M, Shevell M. Genetics and the investigation of developmental delay/intellectual disability. Arch Dis Child. 2014;99:386-9.
- Sagoo GS, Mohammed S. Array CGH testing for learning disability-when is it worth it? En: PHG Foundation [en línea] [consultado el 02/11/2017]. Disponible en: http://www.phgfoundation.org/blog/array-cgh-testing-for-learning-disability-when-is-it-worth-it
- Cigudosa JC, Lapunzina P (coords.). Consenso para la Implementación de los arrays [CGH y SNP-arrays] en la genética clínica. Madrid: Instituto Roche; 2012.
- Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet 2010;14:749-64.
- Park SJ, Jung EH, Ryu RS, Kang HW, Ko JM, Kim HJ, et al. Clinical implementation of whole-genome array CGH as a first-tier test in 5080 pre and postnatal cases. Mol Cytogenet. 2011;4:12-2.
- Natera de Benito D, Póo P, Gean E, Vicente-Villa A, García-Cazorla A, Fons-Estupiña MC. Mosaicismo diploide/triploide: un fenotipo variable, pero característico. Rev Neurol. 2014;59:158-63.
- Smith K, Chandler K, Hindley D, Ramsden SC. Fragile X syndrome testing in the North West. Arch Dis Child. 2013;98:239.
- Summary report of workshop to develop UKGTN testing criteria for fragile x syndrome. En: NHS. UK Genetic Testing Network [en línea] [consultado el 02/11/2017]. Disponible en: https://ukgtn.nhs.uk/resources/library/article/summary-report-of-workshop-to-develop-ukgtn-testing-criteria-for-fragile-x-syndrome-104/
- MacPherson J, Sharif A. Practice Guidelines for Molecular Diagnosis of Fragile X Syndrome. En: Association for Clinical Genetic Science [en línea] [consultado el 02/11/2017]. Disponible en: http://www.acgs.uk.com/media/908997/frx_bpg_final_nov_2014.pdf
- Sherr EH, Michelson DJ, Shevell MI, Moeschler JB, Gropman AL, Ashwal S. Neurodevelopmental disorders and genetic testing: current approaches and future advances. Ann Neurol. 2013;74:164-70.
- Shevell M, Ashwal S, Donley D, Flint J, Gingold M, Hirtz D, et al. Practice parameter: evaluation of the child with global developmental delay: report of the Quality Standards Subcommittee of the American Academy of Neurology and The Practice Committee of the Child Neurology Society. Neurology. 2003;60:367-80.
