Sumario
Uso racional de las pruebas diagnósticas
2013, vol. 6, nº 3

 El electroencefalograma en el estudio y control de la epilepsia
El electroencefalograma en el estudio y control de la epilepsia
Autores: López Pisón J1, Dolz Zaera I2, Arana Navarro T3
1 Pediatra Atención Hospitalaria. Sección Neuropediatría. Hospital Miguel Servet. . Zaragoza (España).
2 Pediatra Atención Hospitalaria. Servicio Neurofisiolófia. Hospital Universitario Miguel Servet. Zaragoza (España).
3 Pediatra Atención Primaria. Pediatra. CS Sagasta. Zaragoza (España).
1 Pediatra Atención Hospitalaria. Sección Neuropediatría. Hospital Miguel Servet. . Zaragoza (España).
2 Pediatra Atención Hospitalaria. Servicio Neurofisiolófia. Hospital Universitario Miguel Servet. Zaragoza (España).
3 Pediatra Atención Primaria. Pediatra. CS Sagasta. Zaragoza (España).
INTRODUCCIÓN
¿Qué es?
El electroencefalograma (EEG) es el registro de la actividad eléctrica cerebral.
¿Cuándo está indicado?
El EEG es útil en:
-
Estudio de la epilepsia:
- Identificación de alteraciones focales o generalizadas que orienten o establezcan el diagnóstico de epilepsia.
- Diagnóstico de un síndrome epiléptico preciso, como síndrome de West/espasmos infantiles, epilepsia ausencias o epilepsia mioclónica juvenil.
- Evidencia de crisis electroclínicas. Puede ser muy útil, pues establece el diagnóstico de epilepsia, en casos de EEG intercríticos normales, lo que es bastante frecuente, especialmente en lactantes, más si las crisis son dudosas. Permite diferenciarlas de trastornos paroxísticos no epilépticos (TPNE), lo que en algunos casos puede ser muy difícil solo con datos clínicos.
- Estudio de encefalopatía aguda, permitiendo identificar estado epiléptico no convulsivo, u orientar hacia algunas intoxicaciones o hiperamoniemia.
- Control del paciente en coma en Unidad de Cuidados Intensivos Pediátricos (UCIP).
- Establecimiento y control del estado eléctrico de brote-supresión, que se busca en el tratamiento del estado epiléptico refractario mediante coma barbitúrico, o midazolam o propofol en dosis altas; estos fármacos disminuyen la actividad eléctrica cerebral en razón directa a la dosis, llegando incluso a EEG isoeléctrico, similar al encontrado en la muerte encefálica.
- Diagnóstico de muerte cerebral, en paciente sometido a ventilación mecánica. Necesario legalmente para el transplante de órganos.
- El polisomnograma (PSNG) nocturno (EEG de sueño nocturno) es útil en el estudio de las parasomnias y su diferenciación con crisis epilépticas y también en la identificación del síndrome de apnea obstructiva del sueño (SAOS). Una limitación es que el niño no duerme igual, con la misma profundidad, en el hospital ni, en casos de registro domiciliario, en su casa con los electrodos colocados.
En este capítulo solo se tratará el EEG en el estudio de la epilepsia, especialmente en edades pediátricas. Revisaremos conceptos básicos de epilepsia y de crisis febriles, que es la causa más frecuente de convulsiones en Pediatría.
CRISIS EPILÉPTICA
La crisis epiléptica es una manifestación muy frecuente de encefalopatía, y supone un motivo muy frecuente de consulta en Pediatría.
Una crisis epiléptica es un trastorno episódico producido por un mecanismo neurofisiológico específico: una anormal actividad ocasional y repetitiva de la sustancia gris cerebral.
Es una manifestación posible en toda encefalopatía aguda o crónica que afecta al córtex cerebral, ya sea de forma primaria como en un traumatismo craneoencefálico o en una encefalitis aguda, o bien como consecuencia cerebral secundaria a diversas patologías como una hipoglucemia o una crisis hipertensiva.
Las expresiones clínicas de las crisis epilépticas son muy variadas y dependen básicamente de la localización cerebral de la descarga anómala. Las crisis parciales, dependiendo de la localización de la descarga anómala, pueden ser de sintomatología motora, sensitiva, sensorial, vegetativa, psíquica o combinación de ellas. Cuando se altera el estado de conciencia se llaman complejas. Pueden generalizar secundariamente. Las crisis generalizadas pueden manifestarse como alteración del estado de conciencia, ausencias, o por combinaciones variables de alteraciones del tono muscular y movimientos anómalos de extremidades.
EPILEPSIA
La epilepsia viene definida por la repetición de crisis epilépticas, excluidas las crisis sintomáticas agudas, como en hipoglucemias o en crisis hipertensivas.
La epilepsia no es un síntoma, ni una enfermedad, ni un síndrome concreto. Es la repetición de una manifestación, la crisis epiléptica, que se puede presentar en prácticamente toda encefalopatía estática o progresiva. Por tanto, los cuadros clínico-evolutivos son muy dispares, dependientes básicamente de la encefalopatía causal. Con el diagnóstico de epilepsia estamos estableciendo el diagnóstico de encefalopatía que ha presentado crisis epilépticas y cuya evolución natural va a ser volver a presentarlas.
El diagnóstico de epilepsia se establece por la repetición de crisis epilépticas. En ocasiones, el EEG puede apoyar o confirmar el diagnóstico.
Hay muchas causas de epilepsia:
- Son frecuentes las epilepsias debidas a alteraciones genéticas cuyo único problema en general es la repetición de crisis epilépticas: epilepsia-ausencias, epilepsia mioclónica juvenil, epilepsia frontal nocturna autosómica dominante o la epilepsia rolándica benigna. Algunas epilepsias genéticas son propias de unas edades concretas, y de la misma manera que un día empiezan, un día acaban. Hay epilepsias genéticas muy graves como el síndrome de Dravet o la epilepsia mioclónica severa del lactante.
- Las epilepsias también pueden ser sintomáticas de muy diversas encefalopatías estáticas o progresivas.
En general, en el estudio de una epilepsia, aparte de una minuciosa anamnesis, especialmente de los episodios, y si es posible la visualización de los mismos, mediante vídeo, solo se precisa estudio EEG y neuroimagen, preferentemente resonancia magnética (RM) cerebral.
No es raro que el diagnóstico de epilepsia sea dudoso, dada la inexistencia de un marcador biológico de certeza.
Epilepsia en la infancia
La epilepsia en el niño tiene unas peculiaridades relacionadas con la edad, lo que justifica diferentes susceptibilidades y diferentes manifestaciones a distintas edades. El tipo de convulsiones a distintas edades viene determinado por el grado de maduración del sistema nervioso central (SNC) y por las variaciones genéticas que influyen en la estructura y bioquímica del cerebro en desarrollo. La edad-dependencia explica por qué las encefalopatías epilépticas clásicas se presentan a edades concretas y por qué un mismo paciente puede evolucionar de una encefalopatía epiléptica precoz a un síndrome de Lennox, pasando por un síndrome de West. Los tres síndromes tienen importantes similitudes clínicas y EEG que apoyan un origen común y representan estadios sucesivos en la maduración de una encefalopatía epiléptica relacionada con la edad. También explica por qué una epilepsia-ausencias de la infancia no se presenta antes de los tres años de edad, o por qué una epilepsia rolándica benigna de la infancia (EBI-R) se presenta y remite espontáneamente a unas edades concretas.
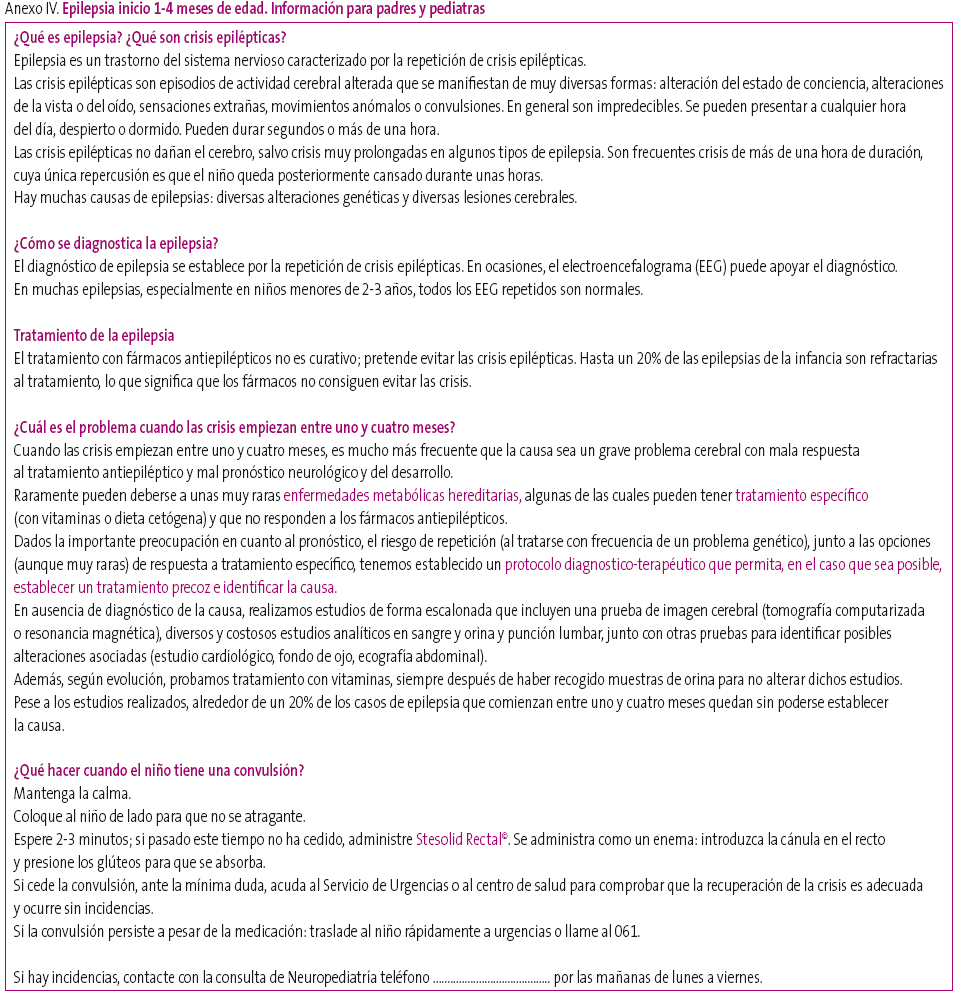
En el Hospital Infantil Miguel Servet de Zaragoza damos una hoja de información a las familias de niños epilépticos o posiblemente epilépticos, con la condición de que tengan hecha o pedida neuroimagen (Anexo I). También damos Hojas de información de epilepsia-ausencias (Anexo II), síndrome de West/espasmos infantiles (Anexo III), epilepsia de inicio entre uno y cuatro meses (Anexo IV) y de crisis febriles (Anexo V). Creemos que son útiles para la comprensión del problema por parte de los familiares y también de profesionales, incluidos los pediatras de Atención Primaria.
CRISIS FEBRILES
Una crisis febril es una crisis convulsiva asociada a una enfermedad febril, en ausencia de una infección del SNC o de un desequilibrio electrolítico, sin antecedente de convulsiones afebriles previas (International League against Epilepsy, Guidelines for epidemiologic studies on epilepsy, 1993).Por regla general, los límites de edad aceptados para la presentación de crisis febriles son los seis meses y los seis años, siendo la máxima incidencia a los 18 meses.
Las crisis febriles son el trastorno convulsivo más frecuente en la infancia, con una prevalencia de un 2-5% en los menores de cinco años.
Las crisis febriles tienen una herencia poligénica multifactorial y puede haber algún subgrupo con herencia autosómica dominante con un patrón de penetrancia reducido.
Se ha descrito que el 21% de las crisis febriles ocurre en la primera hora del proceso febril, el 57% dentro de las primeras 24 horas y un 24% después de las 24 horas. En un 25% de los casos, la crisis es la primera manifestación clínica del proceso infeccioso que la desencadena.
No se ha demostrado que las crisis febriles simples causen daño estructural, alteraciones cognitivas o neuropsicológicas.
Se habla de crisis febril compleja, atípica o complicada ante las siguientes características: duración superior a 15 minutos, crisis focal con o sin generalización secundaria, recurrencia dentro de las primeras 24 horas(dos o más episodios), o hallazgos neurológicos focales en el periodo postcrítico, en un niño sin antecedentes de patología neurológica previa, anormalidad del SNC o historia anterior de crisis afebriles.
El diagnóstico diferencial de las crisis febriles debe realizarse con:
- Crisis convulsivas sintomáticas agudas: infecciones del SNC (meningitis, encefalitis…), convulsiones asociadas a gastroenteritis aguda, especialmente por rotavirus (pueden o no asociar fiebre; es frecuente que se repitan varias en el mismo proceso febril), convulsiones asociadas a procesos respiratorios (pueden o no asociar fiebre) y otras crisis convulsivas sintomáticas agudas (hipoglucemias, otros trastornos hidroelectrolíticos…).
- Epilepsias: la primera manifestación de una epilepsia puede ser una crisis en contexto de fiebre (considerada por tanto como crisis febril, siguiendo la definición de la ILAE).
- Otros trastornos paroxísticos como síncopes febriles o crisis anóxicas febriles, y delirio febril, escalofríos o estremecimientos febriles.
No está indicado ningún estudio ante crisis febriles simples. Ante crisis febriles complejas puede ser aconsejable la realización de un EEG y debe valorarse en algunos casos la realización de neuroimagen.
El riesgo de recurrencia tras un primer episodio de crisis febril varía con la edad: en niños <1 año, 50% de probabilidad de recurrencia, y en niños >1 año, 30% de probabilidad de recurrencia. En los que ya han presentado más de un episodio, el riesgo de una nueva crisis es del 50%. Más de la mitad de las recidivas ocurren entre los 6 y los 12 meses siguientes a la primera crisis.
Los pacientes que han presentado una crisis febril simple tienen un riesgo entre el 1 y el 2,6% de desarrollar epilepsia (frente al 1% de la población general), mientras que los pacientes con crisis febriles complejas pueden desarrollarla en el 4-6% de los casos. El riesgo aumenta en caso de crisis febriles recurrentes, antecedentes de familiares de epilepsia, crisis febriles complejas, duración superior a los 15 minutos, fiebre baja en el momento de la crisis, existencia de anomalías neurológicas previas y primera crisis con una edad menor a los 12 meses.
EPILEPSIA MIOCLÓNICA GAVE DE LA INFANCIA O SÍNDROME DE DRAVET
Es una epilepsia refractaria al tratamiento que asocia deterioro cognitivo. Debe sospecharse ante crisis febriles de inicio antes de los seis meses, estatus febriles generalizados o hemicorporales, muy frecuente repetición de crisis febriles, aparición de crisis afebriles y persistencia de crisis febriles tras los seis años. Los EEG intercríticos iniciales, y en algunos casos durante toda la evolución, suelen ser normales. En los casos más graves de síndrome de Dravet se encuentran mutaciones en el gen SCN1A en alrededor del 70% de los casos. Se habla de espectro de síndrome de Dravet, que incluye cuadros más leves como la epilepsia generalizada con crisis febriles plus (mutaciones en el gen SCN1A en alrededor del 10% de los casos). Su diagnóstico precoz es importante para evitar estudios innecesarios e incertidumbres y para establecer el tratamiento adecuado, pues la evolución es mejor con valproato, topiramato y clobazam, y suelen empeorarla carbamazepina, lamotrigina, vigabatrina y fenitoína. Un cuadro similar en niñas obedece a mutaciones en el gen PCDH19.
ELECTROENCEFALOGRAMA
El EEG tiene las siguientes características:
- Ritmo de base. En adultos, y en niños a partir de los siete años, el ritmo normal en vigilia en derivadas occipitales es α (8-12 ciclos por segundo). En niños, el ritmo de base es más lento, más cuanto más inmaduros.
- Diferenciación en diferentes zonas del cerebro.
- Simetría de las ondas en las diferentes localizaciones en ambos hemisferios. Una rápida visualización permite apreciar la diferenciación y simetría entre el registro en áreas anteriores, medias o posteriores.
- Reactividad a la apertura y cierre de ojos, lo que se valora en la zona occipital.
El registro del sueño cambia con características precisas en las diferentes fases del sueño, empezando en somnolencia por una lentificación generalizada del trazado.
Lo ideal es registrar vigilia y sueño (al menos hasta la fase II de sueño lento), pues hay alteraciones en el EEG, y también crisis epilépticas, que solo aparecen en diferentes fases del sueño, desde la somnolencia inicial hasta el despertar.
Los EEG deben realizarse con hiperpnea (HPN) y estimulación luminosa intermitente (ELI), porque pueden facilitar la presencia de alteraciones en el EEG, y también desencadenar crisis, en casos de algunos síndromes epilépticos.
Hay alteraciones en el EEG muy características de epilepsia:
- Descargas de punta onda (PO), focales o generalizadas. Si se dan generalizadas a tres ciclos por segundo son típicas de epilepsia-ausencias, y con frecuencia se acompañan de alteración del nivel de conciencia. Si son más lentas de tres ciclos por segundo se habla de punta onda lenta (POL), de peor significado pronóstico que la PO a tres ciclos por segundo de la epilepsia-ausencias.
- Descargas de polipunta onda (PPO), habitualmente generalizadas.
- Alteraciones focales persistentes: PO, puntas u ondas agudas.
- Trazado de paroxismos-supresión de las encefalopatías epilépticas precoces.
- Trazado hipsarritmico, muy desestructurado, del síndrome de West.
El trazado EEG, tanto de vigilia como de sueño, cambia con la edad. Un neonato no tiene establecidas las fases del sueño y el ritmo de base es muy lento. Las alteraciones en el EEG también evolucionan. Un neonato no tiene descargas de PO o PPO generalizadas y un niño mayor no puede tener un trazado de paroxismos-supresión ni una hipsarritmia.
Existen factores genéticos determinantes de las características generales del EEG, como la frecuencia y la distribución de la actividad de fondo, y de patrones específicos como la PO generalizada y la respuesta fotoparoxística de descargas generalizadas ante la ELI, que se presentan en determinados intervalos de edad.
EEG en epilepsia
La identificación de alteraciones en la actividad eléctrica cerebral puede ayudar a establecer o a apoyar el diagnóstico de epilepsia y a delimitar el síndrome epiléptico de que se trate. Salvo en raros casos de síndrome de Landau o epilepsia-afasia y el estado de mal epiléptico durante el sueño lento, el diagnóstico de epilepsia exige la existencia de crisis epilépticas.
El EEG es útil para:
- Confirmar la sospecha clínica de un síndrome epiléptico preciso, y documentar el caso: como en epilepsia-ausencias con alteración de conciencia coincidente con descarga de PO generalizada a tres ciclos por segundo, o el trazado hipsarritmico en niños con espasmos en salvas.
- Establecer el diagnóstico de epilepsia en casos dudosos, mediante el registro de crisis electroclínica.
- Apoyar el diagnóstico en casos dudosos, por la presencia de alteraciones de tipo epiléptogeno: PO y PPO generalizadas o persistentes alteraciones focales (PO, puntas u ondas agudas).
- Delimitar el síndrome epiléptico.
Un EEG anormal no implica el diagnóstico de epilepsia
Es conocida la existencia de un patrón de PO generalizada o de puntas rolándicas en personas asintomáticas, especialmente en familiares de pacientes con alguna forma de epilepsia genética como la epilepsia mioclónica juvenil benigna, la epilepsia-ausencias o la epilepsia rolándica benigna de la infancia. La PO generalizada también aparece en el seguimiento de niños con crisis febriles que no desarrollan epilepsia. Otras alteraciones, como PPO generalizada y puntas focales y multifocales se dan durante los años escolares y desaparecen durante esos mismos años o durante la adolescencia en niños que nunca desarrollarán epilepsia. Estas alteraciones generalizadas en el EEG obligan a un estrecho seguimiento, dado que lo más frecuente es que asocien epilepsia antes o después.
Un EEG normal no excluye el diagnóstico de epilepsia
Una persona epiléptica puede tener un registro intercrítico de la actividad eléctrica cerebral normal por existir poca actividad anómala intercrítica o por tratarse de un foco profundo, especialmente en la fosa temporal, no detectable por los electrodos colocados superficialmente en el cuero cabelludo. En muchas epilepsias, especialmente en lactantes, todos los EEG intercríticos repetidos son normales.
Indicaciones del EEG fuera de encefalopatía aguda o paciente en la UCIP
- Estudio de epilepsia o posible epilepsia: diagnóstico diferencial de TPNE. Dado que es frecuente que los EEG intercríticos sean normales, en algunos casos dudosos se debe insistir en el EEG, y puede ser muy útil el registro domiciliario hasta poder registrarse uno de los episodios.
- Confirmación y documentación de determinados síndromes epilépticos. En algunos casos debe hacerse lo antes posible. En el síndrome de West, el pronóstico es mejor (en los casos que van a ir bien) cuanto antes cesen las crisis y se normalice el EEG. Por tanto, el inicio de tratamiento debe considerarse como una urgencia médica, y precisa idóneamente un registro EEG previo. En la epilepsia-ausencias conviene iniciar el tratamiento lo antes posible, tras documentar las ausencias electroclínicas, y controlar estrechamente hasta el cese de las ausencias.
- El PSNGnocturno en el estudio de las parasomnias y su diferenciación con crisis epilépticas y también en la identificación del SAOS.

BIBLIOGRAFÍA RECOMENDADA
- Anderson VE, Hauser WA. Genetics. En: Laidlaw J, Richens A, Chadwick D (eds.). A Textbook of Epilepsy, 4.ª ed. Churchill Livingstone; 1993. p. 47-75.
- Cavazzuti GB, Capella L, Nalin A. Longitudinal Study of Epileptiform EEG Patterns in Normal Children. Epilepsia. 1980;21:43-55.
- Clarke M, Gill J, Noronha M, McKrinlay Y. Early infantile epileptic encephalophaty with suppression-burst: Ohtahara syndrome. Dev Med Child Neurol. 1987;29:520-8.
- Commission on Classification and Terminology of the International League against Epilepsy. Proposal for revised classification of epilepsy and epileptic syndromes. Epilepsia. 1989;30:389-99.
- Delgado-Escueta AV, Serratosa JM. Mapping Epilepsy Genes: Impact on Classification and Genetics Counseling. Rev Neurol (Bar). 1994;114:92-105.
- Donat JF. The Age-Dependent Epileptic Encephalopathies. J Child Neurol. 1992;7:7-22.
- Doose H, Waltz ST. Photosensitivity-Genetics and Clinical Significance. Neuropediatrics. 1993;24:249-55.
- Dravet C, Bureau M, Roger J. L'Epilepsie Myoclonique Bénigne du Nourrison. En: Roger J, Dravet C, Bureau M, Dreifuss FE, Wolf P (eds.). Les syndromes epileptiques de l'enfant et de l'adolescent. John Libbey Eurotext; 1984. p. 51-7.
- Hauser WH, Annegers JF, Anderson VE, Kurland LT. The risk of seizure disorders among relatives of children with febrile convulsions. Neurology. 1985;35:1268-73.
- Van der Meij W, Van Huffelen AC, Willemse J, Schenk-Rootlieb AJF, Meiners LC. Rolandic Spikes in the Inter-ictal EEG pf Children: Contribution to Diagnosis, Classification and Prognosis of Epilepsy. Dev Med Child Neurol. 1992;34:893-903.
Anexo I. Mostrar/ocultar

Anexo II. Mostrar/ocultar

Anexo III. Mostrar/ocultar

Anexo IV. Mostrar/ocultar
Anexo V. Mostrar/ocultar

