Sumario
Puesta al día en...
2015, vol. 8, nº 1

 ¿Qué debe saber el pediatra de Atención Primaria sobre la artritis idiopática juvenil?
¿Qué debe saber el pediatra de Atención Primaria sobre la artritis idiopática juvenil?
Autores: Martínez de Zabarte Fernández J. M1, Lahílla Cuello L2, Medrano San Ildelfonso M3, Arnal Alonso J4
1 MIR-Pediatría. Hospital Infantil Miguel Servet. Zaragoza (España).
2 MIR-Pediatría. Hospital Infantil Miguel Servet. Zaragoza. (España).
3 Servicio de Reumatología. Hospital Miguel Servet. Zaragoza. (España).
4 Pediatra. CS Actur Norte. Zaragoza. (España).
1 MIR-Pediatría. Hospital Infantil Miguel Servet. Zaragoza (España).
2 MIR-Pediatría. Hospital Infantil Miguel Servet. Zaragoza. (España).
3 Servicio de Reumatología. Hospital Miguel Servet. Zaragoza. (España).
4 Pediatra. CS Actur Norte. Zaragoza. (España).
RESUMEN
La artritis idiopática juvenil (AIJ) agrupa a un conjunto de patologías de etiología desconocida y que en su conjunto representan la causa más frecuente de enfermedades reumatológicas en la edad pediátrica. Es importante clasificar al paciente en la categoría diagnóstica que le corresponde ya que el pronóstico y manejo terapéutico son diferentes en cada entidad. Antes de establecerse el diagnóstico de AIJ, debe realizarse un adecuado diagnóstico diferencial con el fin de descartar otras entidades que puedan presentar sintomatología similar. Es importante la valoración y seguimiento por parte de un equipo multidisciplinar de los pacientes con AIJ, ya que deberá seguirse un control intensivo del paciente para prevenir y tratar la enfermedad y las posibles complicaciones. El tratamiento abarca medidas generales, fisioterápicas y un amplio abanico de fármacos analgésicos e inmunosupresores.
INTRODUCCIÓN
La artritis idiopática juvenil (AIJ) es un término establecido desde 1993 por la International League of Associations for Rheumatology (ILAR) para englobar a un grupo heterogéneo de enfermedades que se caracterizan por artritis de causa desconocida de inicio antes de los 16 años de edad y que persiste al menos seis semanas, habiéndose excluido otras causas.
La denominación de AIJ, así como los criterios de diagnóstico están internacionalmente aceptados, sin embargo la clasificación de AIJ está sujeta a un proceso de continua evaluación.
La AIJ es la patología reumática más frecuente en la infancia, con una incidencia en torno a 8-14/105casos/año en menores de 16 años, y una prevalencia de 60-80/105casos/año.
ETIOPATOGENIA
La etiopatogenia de las diferentes formas clínicas que conforman la AIJ es prácticamente desconocida; se considera de origen multifactorial, resultado de reacciones inmunitarias desencadenadas por complejas interacciones de diversos agentes ambientales en un individuo genéticamente susceptible.
Los factores ambientales parecen influir en el inicio y evolución de la enfermedad, siendo la exposición solar, el tabaquismo materno durante el embarazo, las vacunas, la lactancia materna y especialmente las infecciones (parvovirus B19, virus Ebstein-Bar, otros virus y bacterias) los principales agentes desencadenantes del inicio de la enfermedad.
La heterogeneidad de la AIJ determina que posiblemente existan diferentes factores implicados en su desarrollo, así la forma de inicio sistémico cada vez parece más evidente que se comporta como una enfermedad autoinflamatoria en la cual hay una alteración en la respuesta inmune innata, relacionada con determinados polimorfismos en los genes que codifican la IL-6 y el factor de inhibición macrofágica. Se produce una actividad anómala de los fagocitos con gran producción de citoquinas proinflmatorias como IL-1, IL-6, IL-8 y proteínas S-100. Las formas poliarticulares con factor reumatoide positivo (FR+) son el equivalente en la infancia a la artritis reumatoide del adulto y, como esta, presenta anticuerpos anticitrulinados positivos y es frecuente su asociación a HLA-DR4 y HLA-DR1. En las formas oligoarticulares y poliarticulares sin factor reumatoide (FR-) existe un fallo en la regulación de los linfocitos CD4, produciendo un estado proinflamatorio que impide la tolerancia a los autoantígenos y provoca la producción de ANA. Se han descrito diferentes alelos HLA de susceptibilidad para cada una de ellas. En la forma artritis-entesitis, la fuerte asociación con el HLA B27 determina una mayor frecuencia de casos dentro de la misma familia.En la artritis psoriásica se produce un error en la respuesta de los linfocitos CD8, con aumento de TNF-α en líquido sinovial. Se relaciona con polimorfismos del gen que codifica el receptor IL-23.
CLASIFICACIÓN
Cada una de las categorías englobadas en el término de AIJ presenta mecanismos etiopatogénicos diferentes que determinan manifestaciones clínicas, evolución, respuesta al tratamiento y pronóstico muy variable.
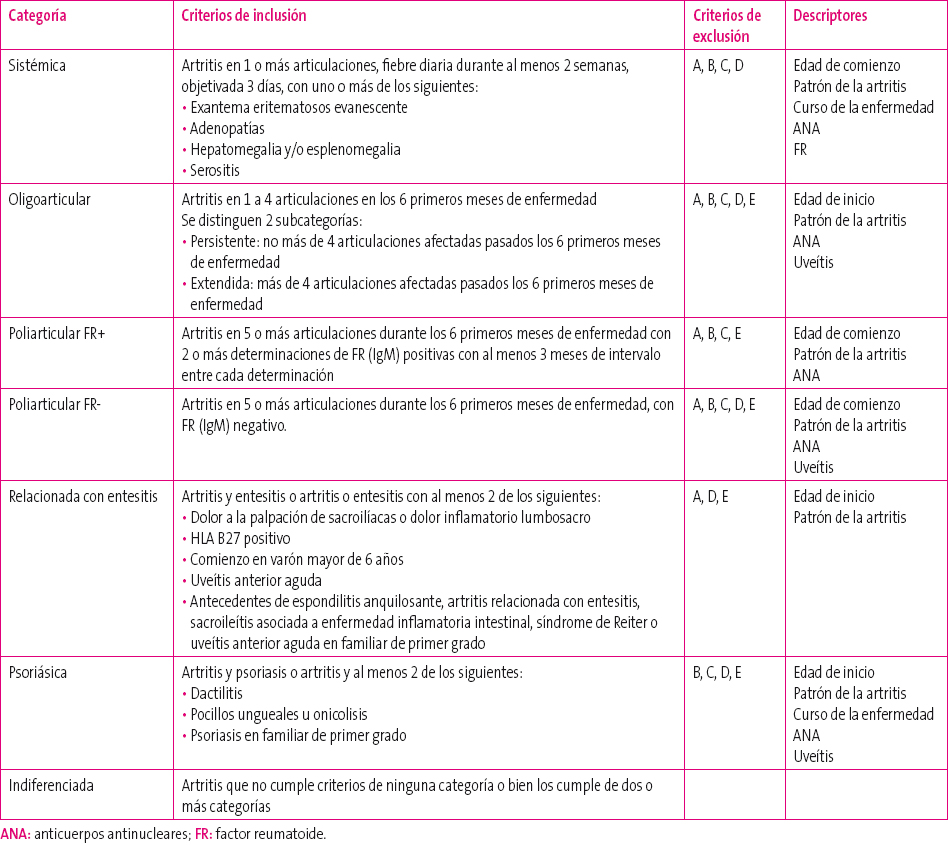
Para la clasificación de las diversas entidades que se engloban en la AIJ se emplean los criterios establecidos en 2001 por la ILAR (Tabla 1). Existen siete categorías con los siguientes criterios de inclusión:
Tabla 1. Categorías diagnósticas de artritis idiopática juvenil (ILAR, 2001). Mostrar/ocultar
-
Sistémica (4-17% de AIJ): artritis en al menos una articulación con fiebre durante dos semanas de forma diaria, con al menos uno de los siguientes:
- Exantema eritematoso evanescente.
- Adenopatías.
- Hepatomegalia y/o esplenomegalia.
- Serositis.
-
Oligoarticular (27-56% de AIJ): artritis que afecta entre una y cuatro articulaciones durante los seis primeros meses de enfermedad. Existen dos subcategorías:
- Persistente: pasados los seis primeros meses de enfermedad, no afecta a más de cuatro articulaciones.
- Extendida: pasados los seis primeros meses de enfermedad afecta a más de cuatro articulaciones.
- Poliarticular FR+ (2-7% de AIJ): artritis que afecta a cinco o más articulaciones durante los seis primeros meses de enfermedad con al menos dos determinaciones de FR (IgM) positivas, existiendo al menos tres meses de intervalo entre cada análisis.
- Poliarticular FR - (11-28% de AIJ): artritis que afecta a cinco o más articulaciones durante los seis primeros meses de enfermedad, con FR (IgM) negativo.
-
Asociada a entesitis (3-11% de AIJ): artritis, entesitis o la combinación de ambas con presencia de al menos dos de los siguientes:
- Dolor a la palpación de sacroilíacas o dolor inflamatorio lumbosacro.
- HLA B27 positivo.
- Comienzo en varón mayor de seis años.
- Uveítis anterior aguda.
- Antecedentes de espondilitis anquilosante, artritis relacionada con entesitis, sacroileítis asociada a enfermedad inflamatoria intestinal, síndrome de Reiter o uveítis anterior aguda en algún familiar de primer grado.
-
Psoriásica (2-11% de AIJ): artritis coexistente con psoriasis o al menos dos de los siguientes:
- Dactilitis.
- Pocillos ungueales u onicolisis.
- Psoriasis en familiar de primer grado.
- Indiferenciada (11-21% de AIJ): artritis que no cumple criterios de ninguna categoría o que los cumple de dos o más categorías.
Además de los criterios de inclusión cada categoría debe cumplir una serie de criterios de exclusión que se han denominado alfabéticamente de la “A” a la “E”:
- Psoriasis o antecedentes de psoriasis en el paciente o en un familiar de primer grado.
- Artritis en un paciente varón, HLA B27 positivo, que sufre la artritis a partir de los seis años de edad.
- Presencia en el paciente o en un familiar de primer grado de: espondiloartritis anquilosante, artritis relacionada con entesitis, sacroileítis con enfermedad inflamatoria intestinal, uveítis anterior aguda.
- FR positivo en al menos dos determinaciones separadas entre sí un mínimo de tres meses.
- Presencia de artritis idiopática juvenil sistémica en el paciente.
- La ILAR, además, enumeró una serie de descriptores para cada una de las categorías, que sin ser criterios de clasificación, son características importantes para cada grupo de pacientes.
MANIFESTACIONES CLÍNICAS, FORMA DE PRESENTACIÓN Y ENFOQUE INICIAL
La sospecha clínica se establece fundamentalmente mediante una correcta anamnesis y exploración del paciente por lo que ante la presencia de un cuadro clínico que haga sospechar alguna de las categorías de AIJ, es importante realizar una anamnesis completa que recoja el tiempo de evolución, episodios previos, intensidad y variación del dolor y otros síntomas, evolución a lo largo del día, dolor nocturno, la presencia de rigidez matutina, limitación en la vida habitual del paciente, contexto o factores con los que aparece la clínica o se agrava e intermitencia de la sintomatología.
Preguntaremos acerca de antecedentes familiares y personales de psoriasis, enfermedades inflamatorias intestinales, espondiloartropatías y otras enfermedades de etiología autoinmune o autoinflamatoria. También se debe obtener información acerca de posibles infecciones pasadas o que puedan presentarse en el momento actual.
En la exploración articular valoraremos en cada articulación la presencia de eritema, calor, tumefacción, signos de traumatismo, número de articulaciones afectas, grado de limitación funcional, analizaremos la marcha del paciente, posición articular en reposo, dismetrías de extremidades, la movilidad activa y pasiva de las articulaciones y la fuerza muscular.
Se debe interrogar acerca de otros síntomas acompañantes: fiebre, adenopatías, pérdida de peso, talalgia, dolor axial, síntomas oculares, digestivos y respiratorios. El exantema en la forma sistémica aparece típicamente con la fiebre, está formado por máculas de 2-5 mm, de predominio en tronco y raíz de extremidades aunque puede afectar a palmas, plantas y cara y es reproducible mediante el fenómeno de Koebner.
Debe tenerse presente que no existen signos ni síntomas patognomónicos de la AIJ.
Siempre que exista un alto grado de sospecha de AIJ debe derivarse a un especialista en reumatología pediátrica para completar el estudio.
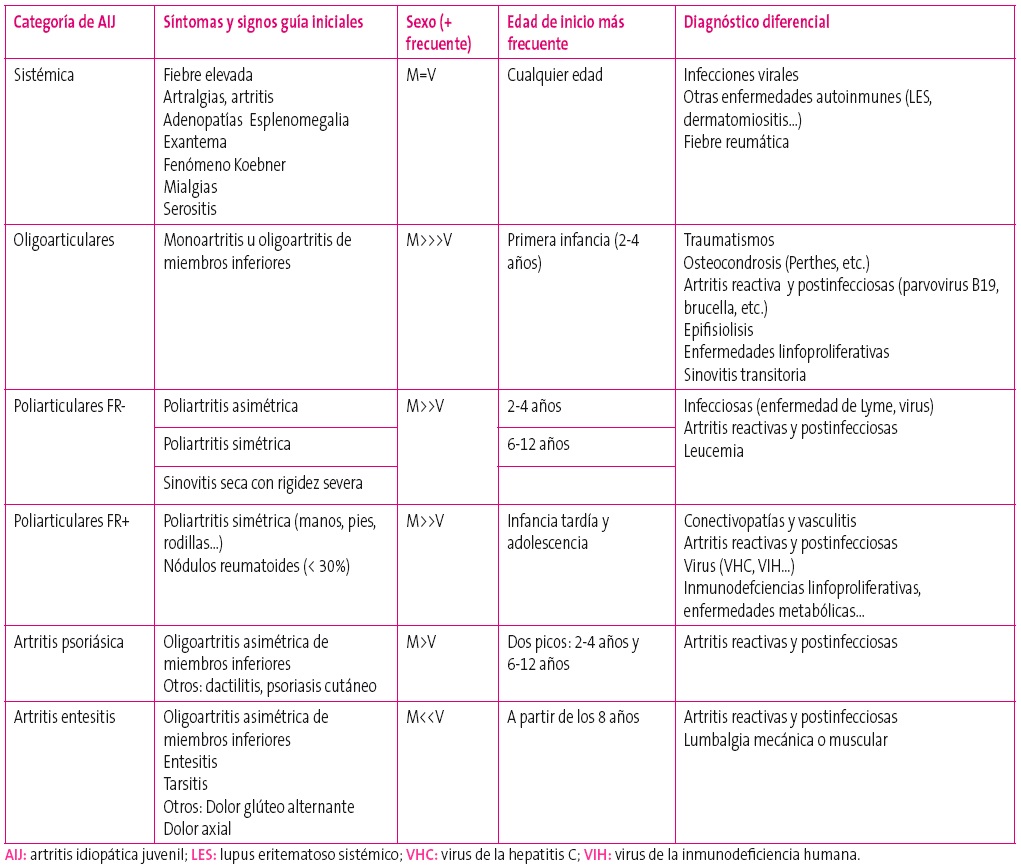
En la Tabla 2 se muestran las principales formas de presentación y algunas entidades con las que debe realizarse el diagnóstico diferencial de las diferentes categorías de AIJ.
Tabla 2. Formas de inicio más frecuentes de las diferentes categorías de artritis idiopática juvenil. Mostrar/ocultar
PRUEBAS COMPLEMENTARIAS
Las pruebas complementarias están dirigidas a realizar un adecuado diagnóstico diferencial, afianzar la sospecha diagnóstica y en algunos casos a confirmarla, ya que forman parte de los criterios diagnósticos. No obstante, el diagnóstico de la AIJ y su clasificación en las diferentes categorías debe establecerse siempre con los criterios previamente explicados.
Ante la sospecha de AIJ es conveniente realizar un análisis de sangre que incluya hemograma y coagulación, bioquímica básica, lactatodeshidrogenasa (LDH), reactantes de fase aguda (VSG, PCR), sedimento de orina y estudio de inmunidad básico: ANA, HLA-B27, inmunoglobulinas, complemento, metabolismo del hierro y marcadores de enfermedad celíaca.
Reactantes de fase aguda (VSG, PCR): se relacionan con el grado de actividad inflamatoria, aunque son inespecíficos. Se elevan en mayor medida en las AIJ sistémica y en las poliarticulares. La VSG puede emplearse para monitorizar la eficacia del tratamiento ya que es un buen marcador de respuesta adecuada al mismo. En la forma artritis-entesitis la PCR suele elevarse en mayor medida que la VSG.
Hemograma: el hallazgo de anemia de trastornos crónicos, leucocitosis con neutrofilia y trombocitosis son frecuentes en la forma sistémica, aunque pueden estar presentes en las otras formas de AIJ. La elevación de la ferritina es un marcador de actividad en la forma sistémica.
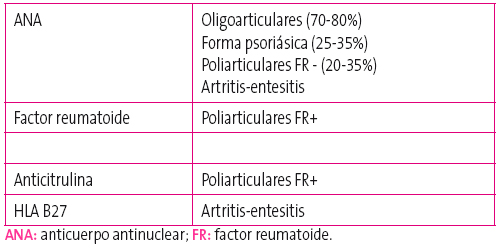
Autoinmunidad y estudio inmunológico: es importante estudiar la presencia de ANA ya que el hallazgo de estos autoanticuerpos en el paciente se relaciona directamente con un mayor riesgo de desarrollar uveítis.
En la Tabla 3 se muestran los hallazgos inmunológicos característicos de cada categoría.
Tabla 3. Estudio de autoinmunidad en las diferentes categorías de artritis idiopática juvenil. Mostrar/ocultar
Estudio microbiológico: en función de la forma de presentación puede ser necesario realizar hemocultivo, serologías para virus de la hepatitis B (VHB), virus de la hepatitis C (VHC), virus de la inmunodeficiencia humana (VIH), virus Ebstein-Bar, parvovirus B19, urocultivo, coprocultivo, frotis faríngeo, Mantoux y cultivo del líquido articular. En todos los casos antes de iniciar un tratamiento con fármacos modificadores de la enfermedad (FAME) se realizarán serología de virus hepáticos, así como Mantoux y booster o quantiferón antes de iniciar el tratamiento biológico.
Estudios de imagen
Radiografía: inicialmente solo observaremos un aumento de partes blandas, posteriormente ensanchamiento articular, crecimiento óseo y osteopenia periarticular, y más tardíamente erosiones y pérdida de alineación.
Ecografía: permite objetivar la presencia, cantidad y características del derrame en la articulación. Puede usarse el Doppler para estudiar la presencia de hipertrofia sinovial y de hiperemia.
Resonancia magnética nuclear (RMN): permite estudiar con mayor detalle las partes blandas y el cartílago, el derrame, las erosiones y el pannus. Se recomienda realizar RMN como prueba de elección en todos los casos HLA-B27 con clínica compatible con sacroileítis, ya que se ha mostrado como una prueba muy eficaz para detectar la presencia de inflamación en estadios iniciales.
Tomografía axial computarizada: útil para estudiar zonas óseas articulares complejas como la zona sacroilíaca cuando la RMN no está indicada. No es recomendable por la alta dosis de radiación.
Gammagrafía: puede encontrarse un aumento de captación en las articulaciones con inflamación activa.
Otras pruebas
Artrocentesis: el líquido sinovial es inflamatorio, de aspecto turbio, en las articulaciones afectas. El análisis puede mostrar aumento de células de serie blanca, glucosa disminuida y proteínas elevadas. Debe cultivarse el líquido para descartar artritis séptica.
Evaluación oftalmológica: debe realizarse en todas las categorías de AIJ para descartar uveítis, pero especialmente cuando existen ANA positivos.
Otras: según la sintomatología del paciente, los años de evolución y la categoría de AIJ que padezca, deberá valorarse la realización de otros estudios: cardiológico, renal, colonoscopia, etc.
PRONÓSTICO Y EVOLUCIÓN
Todas las formas de AIJ pueden producir deformidades en las articulaciones afectas con dismetrías de extremidades y derivar en impotencia funcional si no se controla la enfermedad. Las formas de AIJ con mejor pronóstico son la oligoarticular, que presenta un 50-60% de remisión a los diez años, y la psoriásica, que remite hasta en un 30-50% de los casos. La forma más grave es la sistémica, aunque la mortalidad en nuestro medio se ha reducido a menos del 1%.
En la Tabla 4 se muestran las complicaciones más frecuentes de cada categoría y sus criterios de mal pronóstico.
Tabla 4. Complicaciones y criterios de peor pronóstico en artritis idiopática juvenil Mostrar/ocultar
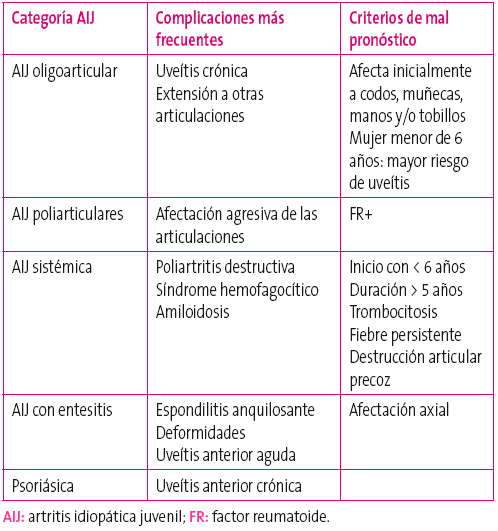
En todas las formas de AIJ, la presencia de anticuerpos antinucleares (ANA+) aumenta el riesgo de desarrollar una uveítis crónica, siendo más frecuente en la forma oligoarticular de inicio antes de seis años y sexo femenino
Es frecuente que existan alteraciones de la mineralización ósea, lo que además de afectar a la estructura del hueso puede alterar el crecimiento del niño, especialmente en la época puberal. En ocasiones la afectación de huesos largos puede estimular una aceleración del crecimiento del mismo, provocando contracturas musculares, dismetrías y escoliosis.
TRATAMIENTO
El tratamiento de los pacientes con artritis idiopática juvenil exige el manejo multidisciplinar de los mismos, de tal manera que se ven implicados los pediatras de atención primaria, el reumatólogo, el oftalmólogo, el traumatólogo, el rehabilitador y el psicólogo pediátrico. Además, el equipo médico debe mantener una buena comunicación con el entorno familiar y escolar del paciente. Solo de esta forma es posible el éxito del tratamiento del paciente en todos los aspectos de la enfermedad: física, mental y socio-familiar.
El objetivo del tratamiento a corto plazo es el alivio del dolor y el control de la actividad inflamatoria (articular y ocular), lo cual permitirá preservar la función articular y prevenir las deformidades. A largo plazo, el objetivo actual es alcanzar la remisión clínica, minimizando los efectos adversos de los medicamentos empleados y promover el desarrollo normal del niño en todas las esferas de su vida.
Medidas terapéuticas farmacológicas
Fármacos disponibles:
- Antiinflamatorios no esteroideos (AINE): se emplean de forma inicial, hasta conseguir la remisión, y cuando existan reagudizaciones o brotes. Suelen repartirse en 2-3 dosis diarias. Los más empleados son: ibuprofeno 25 mg/kg/día, naproxeno 10-20 mg/kg/día e indometacina 2-3 mg/kg/día. Cuando el tratamiento con AINE va a ser prolongado se recomienda asociar un protector gástrico.
- Glucocorticoides: se pueden administrar vía tópica, oral, intramuscular, intraarticular o intravenosa. Debido a su alto perfil de toxicidad deben utilizarse siempre a la menor dosis y el menor tiempo posible. En tratamientos prolongados se recomienda asociar calcio y vitamina D.
-
Fármacos modificadores de la enfermedad (FAME sintéticos):
- Metotrexato: es un análogo estructural del ácido fólico. La dosis estándar es de 10-15 mg/m2. Se puede administrar vía oral o vía subcutánea (mayor biodisponibilidad). Son los más utilizados en la práctica clínica habitual. Los efectos secundarios aparecen en el 15-20% de pacientes y los más frecuentes consisten en náuseas, vómitos, dolor abdominal, aftas, cefalea e irritabilidad. También pueden presentarse anemia y elevación de enzimas hepáticas. Para evitar o disminuir estos efectos secundarios se administra a las 24 horas de la toma del metotrexato una dosis de ácido fólico de 5 mg.
- Otros: leflunomida, sulfasalazina, ciclosporina.
-
Fármacos modificadores de la enfermedad: terapia biológica (FAME biológicos):
-
Antagonistas del factor de necrosis tumoral alfa (TNF-α): el TNF-α es una citocina proinflamatoria secretada por linfocitos T, monocitos, macrófagos y otras células inflamatorias, que induce la activación de los linfocitos y la producción y liberación de otras citocinas.
- Etanercept (Enbrel®): es una proteína de fusión que impide la unión del TNF a sus receptores de superficie celular inhibiendo la actividad biológica del mismo. Se administra por vía subcutánea a dosis de 0,8 mg/kg una vez a la semana. Aprobado a partir de los dos años.
- Infliximab (Remicade®): es un anticuerpo monoclonal quimérico (humano y murino) que se une con alta afinidad tanto a la porción soluble como a la transmembrana del TNF-α inhibiendo su actividad funcional. Se administra por vía intravenosa. No tiene la indicación actual en AIJ, pero se ha mostrado muy eficaz en uveítis.
- Adalimumab (Humira®): es un anticuerpo monoclonal humano recombinante que se une específicamente al TNF-α (tanto al unido a membrana como al soluble) neutralizando su función biológica. Se administra por vía subcutánea cada dos semanas. Aprobado a partir de los dos años.
-
Terapia dirigida frente a linfocitos T:
- Abatacept (Orencia®): es una proteína de fusión que inhibe selectivamente la señal coestimuladora necesaria para la activación de los linfocitos T que expresan CD28. Se administra por vía intravenosa, está indicado a partir de los seis años.
-
Antagonistas de la interleucina 6 (IL-6):
- Tocilizumab (RoActemra®): es un anticuerpo monoclonal IgG-1 humanizado que se dirige tanto al receptor soluble como al de membrana de la IL-6, impidiendo la unión de la IL-6 a sus receptores y bloqueando su acción biológica. Se administra vía intravenosa.
-
Antagonistas de la interleucina 1 (IL-1):
- Anakinra (Kineret®): se administra vía subcutánea de forma diaria. Aunque se emplea en la forma sistémica, no tiene indicación actualmente en Pediatría.
- Canakinumab (Ilaris®): es un anticuerpo monoclonal completamente humano anti-IL-1-β del isotipo IgG-1/κ. Se administra cada ocho semanas mediante inyección subcutánea. Su indicación es en la forma de inicio sistémico.
-
Antagonistas del factor de necrosis tumoral alfa (TNF-α): el TNF-α es una citocina proinflamatoria secretada por linfocitos T, monocitos, macrófagos y otras células inflamatorias, que induce la activación de los linfocitos y la producción y liberación de otras citocinas.
Aproximación terapéutica
El abordaje terapéutico de la artritis idiopática juvenil depende de la forma clínica de la enfermedad, de los factores pronósticos de la misma, de las comorbilidades asociadas y de las características individuales de cada paciente.
Con el fin de homogeneizar al máximo los esquemas terapéuticos, se han publicado múltiples guías para el tratamiento de la AIJ. Así, en 2001, el American College of Rhematology publicó unas recomendaciones para el tratamiento de la AIJ. Estas guías establecen diferentes escenarios clínicos y tratamientos escalonados en cada uno de ellos: artritis de cuatro o menos articulaciones, artritis de cinco o más articulaciones, formas sistémicas con actividad sistémica o articular. Un año más tarde, el grupo Childhood Arthritis and Rheumatology Research Alliance (CARRA) publicó un consenso de tratamiento en el que se establecía el uso escalonado de los diferentes fármacos disponibles en función de la actividad de la enfermedad.
A modo de resumen, en las formas oligoarticulares, el control de la enfermedad debe realizarse con infiltraciones intraarticulares y AINE. En caso de afectación ocular, y debido a la actividad a ese nivel, puede ser preciso el uso de corticoides sistémicos y FAME como el metotrexato. En ausencia de control de la enfermedad, el uso de anti-TNF (adalimumab e infliximab de elección) puede ser necesario.
En las formas oligoarticulares extendidas y poliarticulares, suele ser preciso el uso de FAME como el metotrexato para el control de la enfermedad. Ante el fracaso del mismo se puede valorar un cambio de FAME o el inicio de terapia biológica. En estas categorías, los anti-TNF (etanercept, adalimumab) se han mostrado eficaces, así como los fármacos anti IL-6 (tocilizumab) y anti-linfocitos T (abatacept).
En las formas clínicas con manifestaciones sistémicas el uso de corticoides y la terapia biológica (anti-IL-6 y anti-IL-1) son la base del tratamiento.
Medidas terapéuticas no farmacológicas
Educación sanitaria
Informar a las familias y a los niños.Existen múltiples folletos informativos dirigidos a padres y a niños que podemos proporcionar en la consulta. Poner en contacto a las familias con asociaciones de pacientes también puede resultar beneficioso.
Rehabilitación y fisioterapia
Los pacientes precisarán de un plan individualizado de tratamiento rehabilitador y fisioterápico para evitar rigidez o pérdida de movilidad articular y para recuperar masa muscular y rango de movimiento articular.

BIBLIOGRAFÍA RECOMENDADA
- Calvo Penadés I. Artritis idiopática juvenil de inicio sistémico. Protoc Diagn Ter Pediatr. 2014;1:27-36.
- Inocencio Arocena J, Casado Picón R. Artritis idiopática juvenil poliarticular. Protoc Diagn Ter Pediatr. 2014;1:21-6.
- Inocencio Arocena J, R Casado Picón R. Artritis idiopática juvenil: introducción, criterios de clasificación, mejoría, recaída y remisión. Epidemiología y periodicidad de las revisiones oftalmológicas. Protoc Diagn Ter Pediatr. 2014;1:1-8.
- Marco Puche A, López Montesinos B, Calvo Penadés I. Artritis idiopática juvenil oligoarticular. Protoc Diagn Ter Pediatr. 2014;1:9-19.
- Valero Expósito M, Gámir Gámir ML. Espondiloartritis en la infancia. Protoc Diagn Ter Pediatr. 2014;1:37-47.
