Sumario
Pequeñeces y rarezas
2013, vol. 6, nº 4

 Síndrome del olor a pescado: trimetilaminuria
Síndrome del olor a pescado: trimetilaminuria
PUNTOS CLAVE
- La trimetilaminuria es un raro trastorno del metabolismo, producido por un aumento de la excreción de trimetilamina (TMA) en la orina y otras secreciones corporales.
- La trimetilaminuria primaria se hereda con carácter autosómico recesivo y está causada por un defecto del sistema enzimático flavin monooxigenasa 3 (FMO3).
- Un defecto en la oxidación hepática de la TMA induce su acumulación y eliminación renal, lo que provoca un fuerte olor a pescado en descomposición (podrido).
- Los primeros síntomas aparecen a edad temprana, pero está infradiagnosticada y suele haber un retraso de su detección, por desconocimiento o demora en su consulta.
- El diagnóstico bioquímico precisa de laboratorios especializados. La determinación de una cifra aislada de TMA en orina es insuficiente para el diagnóstico.
- El estudio genético molecular proporciona el diagnóstico definitivo, con el análisis del gen FMO3, que permite detectar las mutaciones asociadas.
- El tratamiento se basa en la disminución controlada de los precursores procedentes de la alimentación, principalmente de su mayor fuente, la TMA N-óxido (TMANO) y la colina.
INTRODUCCIÓN
La trimetilaminuria es un trastorno que se caracteriza por un intenso olor a pescado en descomposición, producido por un aumento en la excreción de un compuesto oloroso, no tóxico y muy volátil, la trimetilamina (TMA), a través de la orina, el aire espirado, el sudor y otras secreciones corporales, que genera un olor muy desagradable, amoniacal y parecido al del pescado podrido, por lo que también se conoce por el nombre de síndrome del olor a pescado1.
Hay dos formas principales descritas, la trimetilaminuria primaria de origen genético,producida por un error congénito de su metabolismo, poco frecuente, que tiene como causa principal un defecto del sistema enzimático flavin monooxigenasa 3 (FMO3), que interviene en la transformación de la trimetilamina, en trimetilamina N-óxido (TMANO), por lo que al fallar su oxidación se acumulan y eliminan cantidades elevadas de TMA, y la forma adquirida,secundaria a enfermedades hepáticas y renales, que cursa con actividad de FMO3 normal. La forma primaria constituye la mayoría de los casos2.
¿CÓMO SE PRODUCE?
La TMA es una amina terciaria alifática sencilla, que se forma en el intestino, por la degradación bacteriana de productos precursores procedentes de la alimentación, principalmente TMANO (componente esencial de los peces marinos, sobre todo del pescado azul), colina, lecitina y carnitina, contenidos en la yema de huevo, las vísceras (hígado, riñón), algunos pescados, en las carnes magras y las verduras (coles, guisantes, judías).
La TMANO y la colina son reducidos en el colon por la microflora intestinal a trimetilamina, que es absorbida por difusión pasiva a través de las membranas celulares, entra en la circulación enterohepática y se elimina por el hígado. En los hepatocitos en condiciones normales, el 95% de las TMA se transforma por acción de la enzima FMO3, en TMANO, compuesto no oloroso y muy soluble en agua, que se excreta principalmente por la orina.Cuando el niño presenta una disminución de la actividad enzimáticapor mutaciones en el gen FMO3,que codifica esta proteína, la transformación de TMA en su forma oxidada no se produce de forma eficaz, o cuando por sobrecrecimiento bacteriano (en la forma secundaria), se genera un aumento excesivo de TMA, la oxidación por la enzima FMO3 no es suficiente, se acumula y se produce su excreción masiva, causando el mal olor corporal3.
Genética de FMO3 y variantes
El gen FMO3 está situado en el brazo largo del cromosoma 1 (q24.3)4. Hasta la fecha, se han registrado más de 300 variantes o polimorfismos de nucleótido simple (SNP) en el gen FMO3. Algunas de ellas han sido identificadas como las mutaciones patógenas causantes de abolir la actividad FMO3, causando trimetilaminuria primaria. Se han descrito al menos 40 mutaciones del gen que se asocian con una mayor o menor actividad de la enzima, lo que hace posible que los individuos que padecen la enfermedad desarrollen formas más o menos graves5. Los casos más severos presentan las mutacionesP153L y E305X4.
Existen formas transitorias, en relación con factores que modifican la capacidad oxidativa de la enzima, como las infecciones víricas (tras una hepatitis viral); la inmadurez del sistema oxidativo, sobre todo en prematuros, lactantes y niños pequeños, que tienen baja la expresión FMO3 o favorecida por una sobrecarga de sustratos; los lactantes alimentados con leches de fórmula que contienen colina; la presencia de inhibidores enzimáticos; el exceso de precursores dietéticos de TMA; los problemas renales o hepáticos; la alteración de la flora intestinal; el incremento de la absorción intestinal y los factores hormonales (episodios favorecidos por la menstruación). Estas formas leves o episódicas suelenaparecer en individuos heterocigotos. Como la trimetilaminuria primaria, se hereda de forma autosómico recesiva (no ligada al sexo), los heterocigotos o portadores de la mutación FMO3 sola son asintomáticos6.
FRECUENCIA
En 1970 Humbert et al.1,2, describieron el primer caso en una niña de seis años de edad, y en la actualidad hay recogidos más de 200 casos en la literatura médica. En España está infradiagnosticada y solo hay publicados cinco hasta la fecha2,7-10, el primero (una niña de cuatro años de edad) fue documentado en la Revista Medicina Clínica, en el año 20032. En la actualidad, la incidencia de trimetilaminuria primaria está en constante revisión, y cada vez es más frecuente la descripción de nuevos casos. La incidencia estimada es de un caso por 40 000 personas, y el porcentaje de heterocigotos en la población general alcanzaría el 1%2,8.
CLÍNICA
Los primeros síntomas aparecen en la infancia, al introducir alimentos ricos en colina y TMANO y con la excepción del mal olor corporal y la halitosis, que pueden acentuarse cuando hay un aumento de la sudoración, fiebre, ejercicio intenso y en situaciones de estrés o de tensión emocional, no hay síntomas que se asocien a la trimetilaminuria. Sin embargo, el síndrome del olor a pescado no puede considerarse un proceso benigno, porque produce un notable deterioro en las relaciones sociales del niño, del adolescente o del adulto, que incide en la escuela, la universidad o el trabajo, y que termina por afectar a la esfera personal. Sus principales consecuencias son generar aislamiento, hacer mella en la autoestima y presentar problemas psicológicos como trastornos del ánimo, episodios de ansiedad, obsesión compulsiva por la higiene corporal, depresión y tendencias suicidas6.
DIAGNÓSTICO
Hasta hace unos años, el diagnóstico se basaba en los síntomas clínicos, más la determinación bioquímica de muestras de orina, de TMA sola o en combinación con su forma oxidada. La prueba de sobrecarga oral, tras la administración de TMA, es útil para identificar a los portadores. Sin embargo, con el advenimiento del genotipo molecular y el reconocimiento de las mutaciones causales, los homocigotos y las formas de portador pueden ser fácilmente reconocidos.
Diagnóstico clínico
Se establece por el olor corporal desagradable que despiden estos pacientes. La TMA excretada es detectada fácilmente por el sistema olfativo humano, en niveles de 0,12 ppb, pero basar el diagnóstico en el sentido del olfato es muy subjetivo, y el mal olor puede ser causado por otros compuestos diferentes. Su presencia también es con frecuencia episódica y puede no ser percibido en el momento de examinar al niño, y aunque el olfato es capaz de discriminar concentraciones tan bajas como una parte por 109, sin embargo, hasta un 7% de las personas normales no son capaces de hacerlo. El diagnóstico clínico es orientativo, permite su sospecha, pero no lo confirma5.
Diagnóstico bioquímico
Una cifra alta aislada de TMA en orina no es suficiente para el diagnóstico, porque puede deberse a una excreción transitoria elevada por sobreproducción, procedente de la dieta o por un aumento de sustratos generales en el intestino. También, al ser un compuesto volátil, en las muestras de orina puede dar lugar a falsos negativos.
Los métodos para identificar la presencia de TMA y su N-óxido en orina precisan de laboratorios especializados y de personal experto, en general no fácilmente accesibles, que incluyan técnicas como cromatografía de gases con espectrometría de masas, para identificar compuestos que producen el mal olor, o la más empleada, la espectroscopia de resonancia nuclear magnética, por su sensibilidad11.
El diagnóstico de trimetilaminuria se basa en el estudio bioquímico de la orina, con la determinación del porcentaje de TMA total (TMA libre más su metabolito N-óxido), excretada en la orina como TMA libre no metabolizada,que en condiciones normales supone hasta un 9%; o de su concentración en orina, expresada en µmol/mmol de creatinina.Se considera trimetilaminuria leve cuando se excreta el 10-39%, y grave, si supera el 40%6,11.
La determinación de la concentración de TMA no metabolizada libre requiere de una prueba de sobrecarga oral, tras haber consumido una dieta normal durante las semanas previas, y la recogida de dos muestras de orina, obtenidas antes y después de haber ingerido pescado fresco marino o alimentos ricos en colina y se cuantifica la concentración de TMA y TMANO mediante espectroscopia de resonancia magnética. La concentración de TMA en muestra de orina basal es menor de 5 µmol/mmol de creatinina (rango de referencia normal), pero en la trimetilaminuria está elevada, junto con concentraciones de TMANO indetectables y tras la sobrecarga de pescado, hay un incremento considerable de ambos. Una concentración urinaria de TMA libre mayor o igual a 10 µg/ml (18-20 µmol/mmol de creatinina) se correlaciona con una excreción de 15-20 mg/día, que parece representar el umbral para la presencia del olor a pescado asociado con el desorden6,11.
Además, los sujetos normales excretan al menos un 90% como TMANO, pero en la trimetilaminuria primaria, la determinación del cociente TMANO/(TMA+TMANO) en orina está muy disminuido en los pacientes homocigotos10.
Detección de portadores
Los familiares heterocigotos (portadores), presentan valores normales de TMA y TMANO en orina y pueden ser identificados con un test de sobrecarga con alimentos precursores. Tras la prueba, los portadores excretan el 20-30% de TMA total como amina libre, mientras que los individuos normales excretan menos del 13%12.
Diagnóstico genético
El diagnóstico de confirmación es complejo y costoso y se hace mediante un estudio de genética molecular, con el análisis de la secuencia de las mutaciones del gen FMO36.
TRATAMIENTO
En la actualidad no hay tratamiento etiológico para la forma genética. El único disponible es el dietético, que se basa en la disminución controlada de los precursores de TMA, procedente de la alimentación, principalmente de su mayor fuente (TMANO y colina). Los niños o sus padres descubrirán los alimentos que aumentan los síntomas. Es importante hacer antes un ensayo, para comprobar que se aminora el mal olor. También puede ser útil el empleo de un calendario dietético durante un corto periodo de tiempo, para determinar los días que presenta olor a pescado, e iniciar el estudio de esta enfermedad10.
Dieta con bajo contenido en precursores2,6,10
TMANO
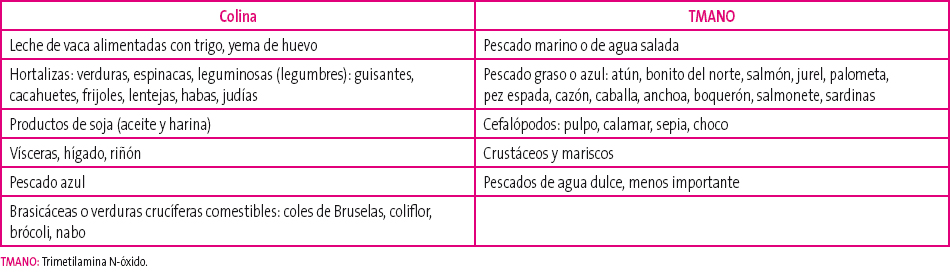
Está presente en pescados de agua salada, especialmente azul, en cefalópodos, como el pulpo o el calamar, y en los crustáceos. Los peces de agua dulce tienen muy bajo contenido y no son un problema.
Colina
Es integrante de diversos alimentos como huevos, vísceras, algunas hortalizas como leguminosas, judías, espinacas o guisantes, frutos secos, productos de soja, algunos pescados (bacalao y salmón), comida rápida (fast food) y hojas y flores del género brassica: coles de Bruselas, coliflor, brócoli... (Tabla 1). Estas también pueden inhibir la actividad de la enzima FMO3 hepática, por su contenido en indoles.
Tabla 1. Alimentos precursores Mostrar/ocultar
La colina es un nutriente esencial muy necesario en una dieta equilibrada, y aunque puede ser sintetizada en menor proporción por metilación de la fosfatidiletanolamina, una deficiencia por dieta estricta puede producir alteraciones importantes como hígado graso, disfunción renal, enfermedad neurológica, retraso del crecimiento, alteraciones óseas y carcinogénesis.
Las necesidades diarias de colina para niños y adolescentes son de 200 a 375 mg, y en adultos sanos, de 425 a 550 mg en mujeres y hombres, respectivamente. La leche materna contiene aproximadamente 1,5 mmol de colina por litro. Un bebé puede consumir 750 μmol, por lo que no se recomienda la restricción estricta de colina de los alimentos en mujeres durante el embarazo o la lactancia, ni en los niños en edad de crecimiento.Un déficit de colina induce el consumo de folato, en la metilación de homocisteína a metionina y una depleción de los depósitos de ácido fólico, por lo que conviene suplementar la dieta con riboflavina (vitamina B2) y ácido fólico, preferentemente a través de la alimentación, (vegetales de hojas verdes o cereales fortificados).
Productos de higiene corporal5
Geles, jabones, lociones y cremas cutáneas de pH ácido (5,5-6,5), para neutralizar químicamente la TMA, que es alcalina. Esta combinación crea una sal menos volátil de trimetilamina, que reduce el olor corporal, puede ser lavada y ayuda a eliminar la TMA secretada.
Disminución del exceso de producción intestinal de TMA
En las formas graves o por razones clínicas o sociales, con2,5,6:
- Antibióticos orales como el metronidazol, administrado de forma ocasional o intermitente y en ciclos cortos de diez días, pueden ayudar a reducir la microflora bacteriana.
- Absorbentes: resinas de intercambio iónico o carbón activado, también en cursos cortos de diez días, para modificar la flora intestinal responsable de la conversión de precursores en TMA.
- Clorofilina de cobre 60 mg tres veces al día después de las comidas, durante tres semanas, para secuestrar la TMA producida en el intestino.
- Laxantes como la lactulosa, para disminuir el tiempo de tránsito intestinal.
No hay hasta la fecha ninguna evaluación sistemática de los diversos tratamientos empleados en el síndrome del olor a pescado. Los intentos de reducir la ingesta de precursores de la dieta, como la TMA, la colina, la lecitina o la carnitina, parecen ser acertados, pero no de manera uniforme en todos los pacientes. Parece evidente que una reducción moderada podría ser más eficaz para mejorar las formas leves y menos graves del síndrome. La sensibilización en su detección, la accesibilidad a las técnicas de diagnóstico y el aumento en la descripción de nuevos casos harán posible en un futuro próximo plantear una revisión del tratamiento. Otras estrategias, como la terapia génica, permitirán reemplazar a corto plazo los genes mutados de FMO3, o la colonización del intestino con microorganismos modificados con FMO3 humana. Los afectados y familiares, pueden beneficiarse del consejo genético si está disponible (Tabla 2).
Tabla 2. Caso clínico. Mostrar/ocultar

BIBLIOGRAFÍA
- Humbert JR, Hammond KB, Hathaway WE,MarcouxJG, O'BrienD.Trimethylaminuria: the fish-odour syndrome. Lancet. 1970;2:770-1.
- Mazón Ramos A, Gil-Setas A, Berrade Zubiri S, Bandrés Echeverri T, Wevers R, Engelke U, et al. Primary trimethylaminuria or fish odor syndrome. A novel mutation in the first documented case in Spain. Med Clin (Barc). 2003;120:219-21.
- Wevers R, Guijarro G. Trimetilaminuria: el síndrome de olor a pescado. Endocrinol Nutr. 2009;56:337-40.
- Hernández D, Addou S, Lee D, Orengo C, Shephard EA, Phillips IR. Trimethylaminuria and a human FMO3 mutation database. Hum Mutat. 2003;22:209-13.
- Mackay RJ, McEntyreCJ, Henderson C, Lever M, George PM. Trimethylaminuria: causes and diagnosis of a socially distressing condition. Clin Biochem Rev. 2011;32(1):33-43.
- Mitchell SC, Smith RL. Trimethylaminuria: the fish malodor syndrome. Drug Metabolism Dispos. 2001;29(4 Pt 2):517-21.
- Fernández MS, Gutiérrez C, Vila JJ, López A, Ibáñez VV, Sangüesa C, et al. Congenital intrahepatic portocaval shunt associated with trimethylaminuria. Pediatr Surg Int. 1997;12:196-97.
- Almenar Bonet MV, Llinares Tello F, Torregrosa Quesada ME, Segrelles Lloret M. Trimetilaminuria (síndrome de olor a pescado): descripción de un caso. Med Clin (Barc). 2008;131:356-57.
- Montoya Álvarez T, Díaz Guardiola P, Olivar Roldán J, Elviro R, Wevers R, Guijarro G. Trimetilaminuria: el síndrome de olor a pescado. Endocrinol Nutr. 2009;56:337-40.
- Romero García A, Bermejo Pastora M, Benito Alonso E, Barros Angueira F, Galán Gómez E. Trimetilaminuria primaria o síndrome del olor a pescado: diagnóstico precoz desde atención primaria. An Pediatr (Barc).2013;78(4):272-3.
- Maschke S, Wahl A, Azaroual N, Boulet O, Crunelle V, Imbenotte M, et al. 1H-NMR analysis of trimethylamina in urine for the diagnosis of fish-odour syndrome. Clin Chim Acta. 1997;263:139-46.
- Al-Waiz M, Ayesh R, Mitchell SC, Idle JR, Smith RL. Trimethylaminuria: the detection of carriers using a trimethylamine load test. J Inherit Metab Dis. 1989;12:80-5.
