Sumario
Situaciones clínicas
2011, vol. 4, nº 2

 Orientación al diagnóstico y tratamiento de las inmunodeficiencias
Orientación al diagnóstico y tratamiento de las inmunodeficiencias
PUNTOS CLAVE
- Las inmunodeficiencias (ID) no son una patología frecuente.
- La importancia del diagnóstico precoz se basa en la prevención de daños crónicos y en alguna entidad, inmunodeficiencia severa combinada (IDSC) se trata de una emergencia médica.
- En más del 50% de los casos un hemograma y una cuantificación de inmunoglobulinas pueden orientar el diagnóstico.
- Una vez orientado el diagnóstico de ID o con alta sospecha del mismo es preciso la consulta con el experto.
- El pediatra debe conocer las modificaciones del calendario vacunal para niños inmunodeficientes, así como medidas generales de profilaxis y tratamiento.
INTRODUCCIÓN
Aunque el diagnóstico de ID corresponde a un laboratorio altamente especializado, en más del 50% de las ID se puede orientar el diagnóstico con una correcta anamnesis y exploración física, un hemograma y una determinación de inmunoglobulinas (Ig) plasmáticas, pruebas disponibles en la mayoría de los laboratorios. En algunas entidades como IDSC y síndrome de delección 22q11 el diagnóstico puede ser una emergencia médica1 por lo que es preciso no demorarlo.
ANAMNESIS
Historia familiar
Hay que preguntar sobre consanguinidad y pertenencia a ciertos grupos étnicos, enfermedades y factores de riesgo durante el embarazo, antecedentes de ID, linfomas, enfermedades autoinmunes y abscesos de repetición diagnosticadas en otros miembros de la familia y muertes precoces no explicadas, (sobre todo en hombres por cuadros con herencia ligada a X)2-4. Pero no siempre hay antecedentes familiares5.
Historia personal
Además de una anamnesis general sobre síntomas generales, hay que recoger datos sobre las infecciones padecidas y características de las mismas, momento de inicio, tipo, número, localización, gravedad, duración, tratamiento y evolución de las mismas, reacciones frente a vacunas y situaciones de anafilaxia ante transfusiones y datos que pudieran orientar a una patología concreta como retraso en desprendimiento del cordón umbilical en déficit de adhesión leucocitaria (LAD)4.
Hay que investigar la posibilidad de ID secundaria y valorar factores de riesgo para infección por virus de la inmunodeficiencia humana (HIV), así como tratamientos inmunosupresores (incluidos corticoides) y enfermedades crónicas.
Se debe registrar la administración de inmunoglobulinas u otros hemoderivados que, además, pueden interferir con el diagnóstico3,4.
Exploración
Una exploración normal no excluye la existencia de inmunodeficiencia.
Puede constatarse la ausencia de tejido linfoide, ausencia de amígdalas y adenopatías no palpables en agammaglobulinemia ligada a X (ALX) o IDSC y hepatoesplenomegalia y adenopatías aumentadas de tamaño en Inmunodeficiencia variable común (IDVC), déficit de IgA e infección por HIV y adenitis supurada en enfermedad granulomatosa crónica (EGC). Se pueden observar secuelas de infecciones previas o patología crónica como acropaquias.
Es frecuente observar en las ID alteraciones cutáneas: eccemas severos, granulomas, vasculitis y otras lesiones autoinmunes.
Puede haber alteración del crecimiento y desarrollo2-4.
En la tabla 1 se muestran los datos de historia y exploración física de interés para el diagnóstico de ID.
Tabla 1. Mostrar/ocultar

Es posible encontrar rasgos dismórficos y datos en la historia que orienten al diagnóstico de algunos síndromes que asocian ID4-7 como los que figuran en la tabla 2.
Tabla 2. Mostrar/ocultar

Pruebas de laboratorio (primer nivel)
Es preciso evaluar los resultados de acuerdo a los valores de referencia para cada edad. (tabla 3).
Tabla 3. Mostrar/ocultar
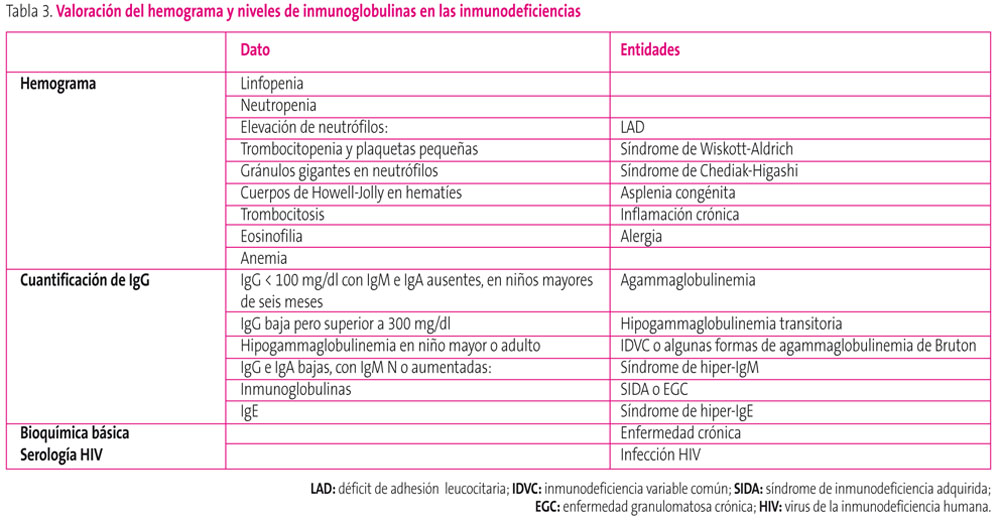
Hemograma
Con recuento de células sanguíneas, que debe hacerse en números absolutos, no en porcentaje. Informa sobre número de linfocitos y neutrófilos así como datos indirectos que orienten a patologías concretas. Puede encontrarse:
- Linfopenia: los valores de linfocitos varían según la edad y el laboratorio. Para algunos autores una cifra inferior a 1500 linfocitos en pacientes mayores de cinco años e inferiores a 2500 en niños menores, es un dato que orienta a ID celular, para otros la cifra sería de menos de 4000-4500 linfocitos/ml en un lactante menor de un año y si es inferior a 2500/ml, debería descartarse IDSC5.
- Neutropenia: cifras inferiores a 1000 en menores de 12 meses y a 1500 en mayores de 12 meses.
-
Elevación en cifra de neutrófilos: debe hacer sospechar LAD. Pueden verse datos indirectos como:
- Trombocitopenia y plaquetas pequeñas en el síndrome de Wiskott-Aldrich.
- Gránulos gigantes en neutrófilos en el síndrome de Chediak-Higashi5.
- Cuerpos de Howell-Jolly en hematíes en asplenia congénita.
- Trombocitosis sugiere inflamación crónica.
- Eosinofilia sugiere alergia.
- Anemia, algunas como la drepanocitosis favorecen las infecciones.
Pueden verse linfopenias y neutropenias transitorias en el seno de infecciones, por lo que estos hallazgos deben ser confirmados y si persisten ser estudiados8.
Bioquímica básica
Con glucosa, función renal, hepática y electrolitos. Informa sobre patología de base y pueden encontrarse datos indirectos que orienten a patologías concretas.
Puede presentarse hiperuricemia en déficit de nucleótido fosforilasa, hipocalcemia en síndrome de DiGeorge, hipoglobulinemias en hipo o agammaglobulinemias e hipoalbuminemia e hipocolesterolemia en malnutrición5.
-
Cuantificación de Ig séricas: informan sobre función humoral. Se deben cuantificar IgG, IgM e IgA para evaluar hipogammaglobulinemia, e IgE.
Si se detecta una hipogammaglobulinemia hay que determinar albúmina para descartar pérdida proteica. Según los valores obtenidos el diagnóstico se puede orientar a3,5:- IgG menor de 100 mg/dl con IgM e IgA ausentes, en niños mayores de seis meses, IgG menor de 200 y suma de IgG, IgA e IgM menor de 400, o ausencia completa de IgM o IgA tras la infancia sugiere agammaglobulinemia congénita.
- IgG baja pero superior a 300 mg/dl, sugiere hipogammaglobulinemia transitoria de la infancia.
- Hipogammaglobulinemia en niño mayor o adulto sugiere IDVC o algunas formas de agammaglobulinemia de Bruton.
- Niveles de IgG e IgA bajos, con IgM normal o alta: síndrome de hiper-IgM.
- Elevación de inmunoglobulinas, hay que descartar EGC o síndrome de inmunodeficiencia adquirida (SIDA).
- Aumento de IgE. Cifras de IgE sobre 100 puede deberse a situaciones de asma, alergia o eccema. Cifras superiores a 2000 sugieren síndrome de Hiper-IgE.
- Cuantificación de subclases de IgG: en pacientes con disminución de IgG total y pobre respuesta de anticuerpos tras vacunación3.
- Serología HIV9: además de bioquímica y hemograma ya mencionados, para excluir inmunodeficiencias secundarias.
- Otros: pueden encontrarse como hallazgos en estudios radiológicos datos que sugieran algunas entidades como ausencia de timo y tejido linfoide, alteraciones costales y vertebrales en déficit de adenosín deaminasa (ADA), alteraciones cardiacas y de grandes vasos en síndrome DiGeorge.
Pruebas de laboratorio especializado3,7
- Medición de anticuerpos específicos contra antígenos proteicos: informa sobre función humoral, mide la funcionalidad de la respuesta frente a tétanos, o polisacáridos como el neumococo. Hay que valorar de acuerdo al estado vacunal del paciente.
- Subclases de linfocitos, y cuantificación de células NK: indicado si se sospecha defecto en células T o B, en linfopenia severa o persistente o presencia de infecciones por oportunistas7. Cualquier alteración de estos resultados debe confirmarse y continuarse de estudios funcionales de esa subclase.
- Hipersensibilidad retardada frente a antígenos a los que el paciente ha sido expuesto previamente mediante una intradermorreacción cuya respuesta se mide a las 48-72 horas. Estudia la función celular.
- Estudios de función humoral in vitro: midiendo producción de inmunoglobulinas frente a mitógenos, citocinas y anti-CD40.
- Estudios de función celular in vitro: valoran la proliferación de células T y la producción de citocinas en respuesta a mitógenos o antígenos específicos.
- Respuesta oxidativa de fagocitos con dihidrorhodamina.
- Defectos de adhesión de leucocitos: CD11 y CD18 ausente en LAD 1 y CD15 en LAD 2.
- Estudio del complemento con CH50 y AP50: en pacientes con sepsis recurrente o por Neisseria. CH50, explora la vía clásica, AP50 la vía alternativa y si una de ellas o ambas están disminuídas se determinan niveles de los factores.
Diagnóstico
El diagnóstico debe completarse con información sobre opciones terapéuticas, pronóstico y consejo genético, por lo que si se confirma alguna patología hay que hacer estudio molecular y genético. En los casos de patologías con herencia recesiva ligada a X hay que identificar a las portadoras.
Es posible realizar diagnóstico prenatal en algunas entidades.
De Vries9 contempla un protocolo diagnóstico escalonado para el diagnóstico de las ID primarias a partir de ocho modelos de presentación clínica, y con el diagnóstico diferencial de cada uno de ellos respecto a otros procesos de etiología no inmune, y Bonilla7 ofrece una revisión exhaustiva de gran parte de los cuadros concretos con algoritmos para el diagnóstico y el manejo terapéutico.
Tratamiento
- Prevención general: mantener aislamiento y ambiente estéril según los casos.
-
Inmunizaciones del niño con ID10:
- Vacuna oral de poliovirus contraindicada en todos los casos por posibilidad de infección diseminada o enfermedad paralítica.
- Vacuna triple vírica, no debe administrarse en las ID celulares, combinadas ni humorales.
- Vacuna de la varicela está contraindicada en ID congénitas, celulares o combinadas, pero no en las humorales y déficits de IgA.
- Las vacunas BCG y rotavirus no deben utilizarse.
- En las deficiencias del sistema mononuclear-fagocítico únicamente esta contraindicada la vacunación con BCG.
- Las vacunas muertas pueden ser administradas en todos los casos tanto en déficit de la inmunidad celular como humoral.
- La vacuna antineumocócica 13-valente está indicada por debajo de los cinco años con pauta de vacunación secuencial con la polisacárida 23-valente a partir de los dos años. Por encima de los cinco años se utilizara solo la 23-valente.
- La vacuna antigripal se debe administrar anualmente en todos los tipos de inmunodeficiencia primarias.
Conviene vacunar a familiares susceptibles con vacuna antivaricela y vacunación antigripal anual.
- Profilaxis específica: profilaxis en ID celulares, frente a Pneumocystis jiroveci con trimetoprim-sulfametoxazol (150 mg/m2 de trimetoprim y 750 mg/m2 de sulfametoxazol) tres días o todos los días a la semana y frente a Aspergillus con itraconazol. En EGC profilaxis de las infecciones bacterianas con trimetoprim sulfametoxazol a las dosis indicadas y profilaxis de las infecciones por Aspergillus con itraconazol5.
- Tratamiento general: tratamiento precoz de las infecciones con antibiótico empírico tras recoger cultivos.
-
Tratamiento específico1,7:
- Trasplante de médula ósea en IDSC y se puede considerar en algunas ID celulares y algunos defectos de fagocitosis.
- Terapia sustitutiva con Inmunoglobulinas intravenosas (IgIV) o subcutánea (IgSC) en Agammaglobulinemia ligada a X y autosómica recesiva (XLA, ARA) o IDVC y en combinadas previas al trasplante. Se pauta cada 3-4 semanas para mantener IgG > 500 o a veces > 800, además del tratamiento de las infecciones.
- En alguna entidad se administran interferón, citocinas y serían susceptibles de terapia génica.
-
Otros:
- Si se necesita transfundir a pacientes con ID de células T, se deben administrar productos irradiados, para evitar enfermedad injerto contra huésped y no utilizar como donantes a familiares que puedan ser potenciales donantes de médula, para evitar sensibilizaciones antigénicas4,5.
- No debe administrarse inmunoglobulina IV hasta estudiar al paciente, puede negativizar un estudio durante meses.
- Control de función pulmonar7.
ALGUNAS ENTIDADES
- Déficit de IgA: es la inmunodeficiencia más frecuente (1/300-700), cursa sin síntomas en la mayoría de casos, aunque puede producir infecciones respiratorias y gastrointestinales recurrentes, también puede asociar enfermedades autoinmunes, como enfermedad celiaca, y las personas que lo padecen pueden presentar reacciones anafilácticas frente a hemoderivados. En algún caso puede progresar a IDVC.
- Inmunodeficiencia variable común (IDVC): hay disminución de cifras de IgG, IgA e IgM y mala respuesta a la inmunización en ausencia de otra ID. Tiene dos picos de frecuencia en la edad de inicio, en preescolares y en la segunda o tercera década de la vida. Los pacientes afectados presentan neumonías, sinusitis, OMA, conjuntivitis, enfermedad crónica pulmonar, bronquiectasias y síntomas gastrointestinales.
- Inmunodeficiencia combinada severa (IDCS): en este cuadro hay ausencia de inmunidad celular y humoral por lo que los pacientes presentan infecciones graves recurrentes por cualquier tipo de gérmenes, virus, hongos, oportunistas… Asocia diarrea crónica, fallo del crecimiento, enfermedad injerto contra huésped y tiene un pronóstico fatal en primer año de vida.
- Déficit de subclases de IgG: déficit de niveles de una o más subclases de IgG. Usualmente es asintomática aunque puede presentarse con infecciones virales o bacterianas recurrentes.
BIBLIOGRAFÍA
- Martín-Nalda A, Soler-Palacín P, Español Borén T, Caragol Urgelles I, Díaz de Heredia Rubio C, Figueras Nadal C. Espectro de las inmunodeficiencias primarias en un hospital de tercer nivel en un periodo de 10 años. An Pediatr (Barc). 2011;74:74-83.
- Slatter MA, Gennery AR. Clinical Immunology Review Series: An approach to the with recurrent infections in chilhood. Clin Exp Immunol. 2008;152(3):389-96.
- Stiehm R. Approach to the child with recurrent infections. Uptodate. Última revisión mayo 2010. www.uptodate.com [consultado el 17-10-2010].
- Ballow M. Approach to the Patient with Recurrent Infections. Clin Rev Allerg Immunol. 2008;34:129-40.
- Ruiz Contreras J. El niño con infecciones frecuentes. En: AEPap ed. Curso de Actualización Pediatría 2010. Madrid: Exlibris Ediciones; 2010.p. 15-22.
- International Union of Immunological Societies Expert Committee on Primary Immunodeficiencies: Notarangelo L, Fischer A, Geha RS, Casanova JL, Chapel H, Conley ME et al. Primary immunodeficiencies: 2009 update. J Allergy Clin Immunol. 2009;124:1161-78.
- Bonilla FA, Bernstein I, Khan DA, Ballas ZK, Chinen J, Frank MM et al. Practice parameter for diagnosis and management of primary immunodeficiency. Ann Allergy Asthma Immunol. 2005;94:S1-64.
- Bonilla FA. Laboratory evaluation of the immune system. Uptodate. Última revision mayo 2010. www.uptodate.com [consultado el 21-10-2010].
- De Vries E. Patient-centred screening for primary immunodeficiency: a multi-stage diagnostic protocol designed for immunologist. Clin Exp Immunol. 2006;145:204-14.
- de Juan Martín F. Vacunación en niños con patología de base. Inmunodeprimidos. Libro de ponencias del 58 Congreso de la Asociación Española de Pediatría. Zaragoza, junio 2009. p. 296-300.
LECTURAS RECOMENDADAS
-
Bonilla FA, Bernstein I, Khan DA, Ballas ZK, Chinen J, Frank MM et al. Practice parameter for diagnosis and management of primary immunodeficiency. Ann Allergy Asthma Immunol. 2005;94:S1-64.
Es una revisión exhaustiva de gran parte de los cuadros concretos con algoritmos para el diagnóstico y el manejo terapéutico. -
De Vries E. Patient-centred screening for primary immunodeficiency: a multi-stage diagnostic protocol designed for immunologist. Clinical and Experimental Immunology. 2005;145:204-14.
Alto interés porque contempla un protocolo diagnóstico escalonado para el diagnóstico de las ID primarias a partir de ocho modelos de presentación clínica, y con el diagnóstico diferencial de cada uno de ellos respecto a otros procesos de etiología no inmune.
